Objectifs pédagogiques
- Argumenter les principales hypothèses diagnostiques et justifier les examens complémentaires pertinents.
- Connaître les principes du traitement de la maladie cœliaque.
Avant de commencer…
L’étiologie des diarrhées chroniques chez l’enfant est habituellement reliée à l’âge.
La démarche diagnostique est surtout clinique, orientant le choix des examens complémentaires.
Les causes les plus fréquentes sont fonctionnelles, notamment chez le jeune enfant. Un retentissement staturo-pondéral signe habituellement l’organicité.
Une diarrhée chronique sans retentissement pondéral est a priori fonctionnelle.
Le tableau typique est l’intestin irritable, première cause chez l’enfant. Certaines maldigestions peuvent cepen-dant permettre une croissance normale ; c’est le cas de l’intolérance physiologique au lactose.
Les points clés à connaître sur la maladie cœliaque et les MICI sont développés ici.
La mucoviscidose fait l’objet d’un chapitre individualisé (voir chapitre 63). Sa présentation sous forme de diar-rhée chronique inexpliquée est devenue rarissime depuis la généralisation du dépistage, mais il faut tout de même savoir l’évoquer compte tenu des faux négatifs possibles.
L’allergie aux protéines du lait de vache est abordée dans le chapitre 59, dans la diversité de ses présentations cliniques, dont la diarrhée chronique.
I. Pour bien comprendre
La diarrhée est caractérisée par l’émission de selles de consistance anormale (molles ou liquides), trop abondantes, trop fréquentes.
Elle est définie comme chronique au-delà de 3 semaines d’évolution.
L’analyse sémiologique est une étape essentielle qui permet d’éviter de toujours faire une pathologie d’une plainte alléguée des parents. En effet, la couleur et la consistance des selles sont physiologique-ment variables. Il convient de savoir reconnaître la fausse diarrhée de l’enfant constipé (association de selles molles et dures, encoprésie), les selles molles/liquides grumeleuses jaunes de l’enfant allaité au sein, les selles vertes lors de l’utilisation de préparations hydrolysées.
L’émission de selles la nuit est un bon argument en faveur d’une diarrhée organique (hormis chez le jeune nourrisson).
II. Argumenter les principales hypothèses diagnostiques et justifier les examens complémentaires pertinents
A. Démarche diagnostique
L’arbre décisionnel (fig. 20.1) présente les principales causes en pédiatrie.
Les causes de diarrhée chronique chez le nouveau-né sont du ressort du spécialiste et ne seront donc pas traitées car il s’agit de pathologies rares.
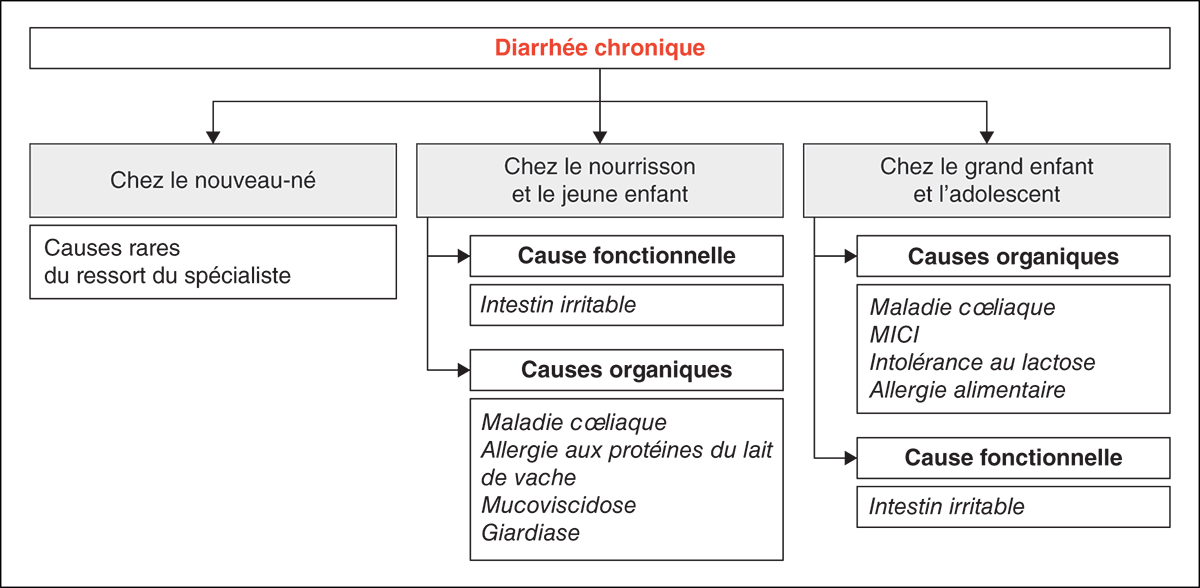
Fig. 20.1. ![]() Principales causes de diarrhée chronique selon l’âge.
Principales causes de diarrhée chronique selon l’âge.
B. Situations cliniques
1. Chez le nourrisson et l’enfant
La diarrhée fonctionnelle associée ou non au syndrome de l’intestin irritable est la cause la plus fréquente.
Elle est caractérisée par une diarrhée abondante nauséabonde isolée, avec débris alimentaires, survenant chez un enfant typiquement âgé de 6 mois à 3 ans, en parfait état général et ayant une croissance normale. La diarrhée est dans ce cas un symptôme isolé.
Aucun examen paraclinique n’est nécessaire au diagnostic. En cas de doute (hyperphagie, selles graisseuses), un dosage de l’élastase fécale peut éliminer une insuffisance pancréatique exocrine (autre cause de diarrhée chronique avec croissance parfois conservée grâce à une hyperphagie compensatrice).
Le traitement fait appel à une modification du régime alimentaire visant à réduire les sucres fermentes-cibles : lactose, fructose, oligosaccharides et polyols (FODMAP). Une surveillance simple est nécessaire car il n’y a pas de complications.
Les causes organiques les plus importantes font l’objet de paragraphes ou chapitres spécifiques : la maladie cœliaque, l’APLV, la mucoviscidose.
L’intolérance au lactose congénitale est rarissime et ne doit donc pas être évoquée aisément chez le nourrisson et le jeune enfant.
2. Chez le grand enfant et l’adolescent
Les MICI sont à évoquer (voir § III.B. Maladies inflammatoires chroniques de l’intestin (MICI)) ainsi que la maladie cœliaque.
Le syndrome de l’intestin irritable se traduit par un tableau clinique identique à celui de l’adulte, avec notamment une croissance staturo-pondérale conservée. Des douleurs abdominales peuvent être associées à de la diarrhée, de la constipation ou une alternance des deux.
La maladie cœliaque peut se révéler à cet âge.
L’intolérance au lactose dans la forme physiologique de l’adulte peut être en cause après l’âge de 5 ans, mais elle est bien plus rare que chez l’adulte. Elle se manifeste par des symptômes (douleurs, ballonne-ment, diarrhée) dans l’heure suivant la prise de lactose.
Les allergies alimentaires sont une cause de diarrhée chronique exceptionnelle à cet âge.
Cause principale chez l’enfant : diarrhée fonctionnelle.
En cas de retentissement pondéral, évoquer :
- chez le jeune nourrisson : APLV, mucoviscidose, maladie cœliaque (si gluten introduit) ;
- chez le préadolescent et l’adolescent : maladie cœliaque, MICI.
III. Points clés à propos de certaines causes
A. Maladie cœliaque
1. Généralités
La maladie cœliaque est une maladie dysimmunitaire déclenchée et entretenue par l’ingestion de gluten chez des sujets génétiquement prédisposés.
Les patients avec maladie cœliaque sont porteurs du génotype HLA-DQ2 (95 %) et/ou DQ8 (5 %). À noter cependant que ce génotype HLA est fréquent, puisque pré-sent chez 30 à 40 % de la population générale, ce qui suggère l’implication d’autres facteurs.
Un membre de la famille atteint de maladie cœliaque augmente le risque.
2. Diagnostic
Enquête clinique
Manifestations cliniques typiques chez le nourrisson diversifié avec du gluten :
- signes digestifs : diarrhée chronique, ballonnement abdominal ;
- cassure pondérale puis staturale ;
- anorexie, dénutrition progressive avec amyotrophie (fig. 20.2) ;
- pâleur, apathie, tristesse.
Les formes atypiques sont en augmentation : carence en fer, retard statural isolé (sans symptômes digestifs et sans cassure pondérale), douleurs abdominales isolées, constipation, vomissements isolés, augmentation des transaminases, hypoplasie de l’émail dentaire, dermatite herpétiforme, hippocratisme digital, retard pubertaire, arthralgies, ostéoporose, aphtes récurrents.
De nombreux contextes doivent faire évoquer l’association possible avec la maladie cœliaque : déficit en IgA, trisomie 21, syndrome de Turner, pathologies auto-immunes dont le diabète de type 1 et la thyroïdite auto-immune.
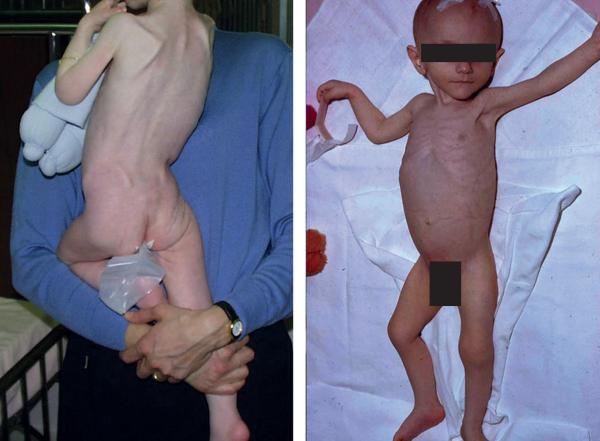
Fig. 20.2. ![]() Enfants dénutris dans un contexte de maladie cœliaque.
Enfants dénutris dans un contexte de maladie cœliaque.
Enquête paraclinique
Le dosage des anticorps est la première étape diagnostique : IgA sériques anti-transglutaminase avec dosage simultané des IgA totales pour éliminer un déficit (5 % des cas en moyenne).
En cas de déficit en IgA, un dosage des IgG anti-transglutaminase pourra être réalisé.
Attention : le dosage des anticorps anti-transglutaminase ne doit être réalisé que chez un patient qui consomme du gluten, son exclusion pouvant faussement négativer le résultat.
Si les IgA anti-transglutaminase sont supérieures à 10 fois la normale, un dosage des IgA anti-endomysium est demandé sur un deuxième prélèvement. La positivité conjointe de ces deux conditions permet de poser le diagnostic de maladie cœliaque par un gastropédiatre sans réaliser de biopsie intestinale.
Dans tous les autres cas, une biopsie intestinale est nécessaire pour poser le diagnostic.
Le diagnostic peut être posé sur les biopsies s’il existe une augmentation des lymphocytes intra-épithéliaux et une hypertrophie des cryptes, la présence d’une atrophie villositaire n’étant pas obligatoire.
Test thérapeutique
La preuve définitive du diagnostic sera apportée par la réponse au régime d’exclusion stricte du gluten. On observe dans ce cas une amélioration :
- clinique : disparition des signes en 1 à 2 semaines, reprise de la croissance (rattrapage staturo-pondéral en 6 à 12 mois) ;
- biologique : négativation des anticorps en 12 à 18 mois le plus souvent, parfois plus.
3. Prise en charge thérapeutique et suivi
Régime d’exclusion
Le régime d’exclusion consiste en l’éviction du gluten (blé, orge, seigle), quel qu’en soit le mode de présentation (plats cuisinés industriels, aliments panés, etc.). La plupart des patients tolèrent de petites quantités d’avoine.
Il peut être couplé initialement et très transitoirement avec un régime sans lactose (notamment durant la phase diarrhéique) et une supplémentation en vitamine D et en fer.
Ce régime d’exclusion doit être poursuivi à vie.
Une éducation thérapeutique, avec conseils diététiques et l’aide d’associations de malades, est utile pour favoriser l’observance et donc l’efficacité du traitement. Le régime est parfois difficile à accepter pour l’enfant et son entourage, notamment en cas de repas en collectivités ou à l’approche de l’adolescence. Un soutien psychologique peut s’avérer utile.
Un PAI est indispensable à l’école, en rassurant les enseignants sur l’absence de risques immédiats liés à une erreur ponctuelle de régime.
La prescription des produits sans gluten peut donner lieu à des remboursements accordés par la Sécuri-té sociale, dans certaines limites et sous certaines conditions (ce n’est pas une maladie ALD-30).
Suivi de l’enfant
L’efficacité et l’observance du régime sont appréciées cliniquement dans les 3 premiers mois et biologi-quement après 12 mois par le dosage des anticorps.
Le mauvais suivi du régime peut s’accompagner chez l’enfant d’un retard de croissance, d’une ostéopé-nie, et d’une augmentation de l’incidence d’autres maladies auto-immunes.
Prédisposition génétique incontournable : HLA-DQ2 ou HLA-DQ8.
Évoquer la maladie cœliaque après l’introduction du gluten chez un nourrisson dénutri avec diarrhée chronique et ballonnement abdominal ou devant des présentations atypiques.
Test diagnostique essentiel et suffisant en première intention : IgA anti-transglutaminases (± IgA totales).
Régime avec éviction du gluten à vie. Ne pas le débuter avant confirmation diagnostique.
B. Maladies inflammatoires chroniques de l’intestin (MICI)
1. Généralités
Les MICI regroupent comme chez l’adulte la maladie de Crohn, la rectocolite hémorragique (RCH) et les colites indéterminées, dont l’incidence est en augmentation chez l’enfant.
Elles peuvent se révéler à tout âge et il est important d’y penser pour éviter un retard diagnostique.
2. Diagnostic
Il convient de savoir avant tout différencier des douleurs abdominales organiques et fonctionnelles (voir chapitre 17). Les signes évocateurs de maladie de Crohn et de RCH sont présentés dans le tableau 20.1.
Tableau 20.1. ![]() Comparaison des signes de maladie de Crohn et RCH chez l’enfant.
Comparaison des signes de maladie de Crohn et RCH chez l’enfant.
| Maladie de Crohn | RCH | |
|---|---|---|
| Diarrhée Sanglante |
Quasi toujours Occasionnelle |
Quasi toujours Quasi toujours |
| Douleurs abdominales | Quasi toujours | Fréquentes |
| Symptomatologie périnéale | Fréquente | Absente |
| Retard de croissance | Fréquent | Rare |
| Dénutrition | Fréquente | Rare |
Des examens complémentaires seront utiles pour orienter vers une MICI, notamment biologique avec recherche d’un syndrome inflammatoire, d’une anémie, d’une hypoalbuminémie, et par imagerie (écho-graphie).
Références
- Husby S, Koletzko S, Korponay-Szabó I, et al. European Society Paediatric Gastroenterology, Hepatology and Nutrition. Guidelines for diagnosing coeliac disease. J Pediatr Gastroenterol Nutr 2020;70:141–56.
- Levine A, Koletzko S, Turner D, et al. ESPGHAN revised Porto criteria for the diagnosis of inflammatory bowel disease in children and adolescents. J Pediatr Gastroenterol Nutr 2014;58:795–806.

|
Lengliné H, Fabre A. Diagnostic de la maladie cœliaque chez l’enfant. Pas à Pas en pédiatrie. Arbres décisionnels commentés des Sociétés de Pédiatrie. 2022. https://pap-pediatrie.fr/files/2022_01_diagnostic_de_la_maladie_coeliaq…; |