Objectifs pédagogiques
- Expliquer les bases du conseil génétique, les possibilités de dépistage et de diagnostic prénatal (modalités et options de prise en charge dans le cadre d’une maladie d’une particulière gravité).
- Expliquer la prescription des tests génétiques : organisation et aspects réglementaires (voir item 9).
- Expliquer les problèmes liés à la maladie et les retentissements de l’arrivée d’un enfant souffrant de maladie génétique sur le couple et la famille.
Avant de commencer…
Le syndrome de l’X fragile est une affection génétique liée à l’X. L’X fragile est la deuxième cause génétique de déficience intellectuelle, après la trisomie 21.
L’anomalie génétique responsable est une mutation instable dans le gène FMR1, qui code une protéine (FMRP) indispensable au fonctionnement synaptique. Le gène normal contient une séquence de plu-sieurs triplets CGG. Cette suite de triplets prédispose à des erreurs lors de la réplication de l’ADN, qui conduisent à une expansion du nombre de triplets. Celle-ci s’aggrave au fil des générations.
-
L’expansion du nombre de triplets CGG est responsable de trois phénotypes pouvant s’observer dans la même famille :
-
la mutation complète est à l’origine d’une déficience intellectuelle dans les deux sexes : le syndrome de l’X fragile ;
-
la prémutation induit deux phénotypes :
-
une maladie neurodégénérative touchant les adultes de plus de 50 ans : le syndrome FXTAS ;
-
l’insuffisance ovarienne avec ménopause prématurée.
-
Le diagnostic clinique d’X fragile repose sur l’association de signes peu spécifiques : déficience intellectuelle associée à des troubles du comportement (notamment traits autistiques), visage allongé avec gros périmètre crânien, grandes oreilles, et macroorchidie après la puberté.
Le diagnostic est confirmé par un test génétique (évaluation du nombre de triplets CGG et de la méthylation du locus FMR1). Un diagnostic prénatal peut être proposé aux couples à risque. En cas de diagnostic d’X fragile, l’information des apparentés à risque est obligatoire.
I. Pour bien comprendre
A. Données épidémiologiques
Avec une prévalence de 1/4 000 garçons et de 1/5 000 à 1/8 000 filles, le syndrome de l’X fragile explique 2 à 3 % de la déficience intellectuelle et représente la première étiologie monogénique et la deuxième cause de déficience intellectuelle après la trisomie 21.
B. Données génétiques
Le gène FMR1 (Fragile X Mental Retardation 1) est situé à l’extrémité du bras long du chromo-some X. Il est transcrit en un ARN messager (ARNm) codant la protéine FMRP. FMRP est une pro-téine chaperonne qui peut se lier à des ARNm. Elle contrôle la synthèse protéique au sein des épines dendritiques, en transportant divers ARNm, dont elle module la traduction, du noyau vers la région présynaptique. Elle est indispensable pour la plasticité synaptique.
Les mutations par expansion de triplets sont une classe particulière de mutation, dites « instables » puisque l’expansion se modifie à chaque génération (tableau 13.1). D’autres pathologies génétiques sont liées à des mutations instables, comme la chorée de Huntington et la dystrophie myotonique de Steinert.
Tableau 13.1. ![]() Caractéristiques moléculaires des mutations de FMR1.
Caractéristiques moléculaires des mutations de FMR1.
| Nombre de triplets CGG | État | Stabilité | Parent transmetteur |
|---|---|---|---|
| 6 à 44 (moyenne : 30) | Normal | Expansion exceptionnelle vers une prémutation | |
| 45 à 54 | Intermédiaire | Expansion rare vers une prémutation, jamais vers une mutation | Par le père (à un fœtus féminin) ou la mère |
| 55 à ≈ 200 sans méthylation | Prémutation | Expansion systématique vers une prémutation plus grande ou vers une mutation | Par le père (à un fœtus féminin) ou la mère |
|
> ≈ 200 avec méthylation |
Mutation complète |
Expansion variable Pas de retour à la prémutation |
Par la mère (prémutée ou mutée) |
Première cause monogénique de déficience intellectuelle.
Mutation complète dans le gène FMR1 (expansion de triplets ≥ 200 CGG avec méthylation du locus).
II. Faire le diagnostic de mutation dans le gène FMR1
A. Tableau clinique du syndrome de l’X fragile
1. Généralités
Prémutation et mutation du gène FMR1 sont responsables de maladies différentes, par deux mé-canismes physiopathologiques distincts : toxicité nucléaire des ARNm pour la prémutation, ab-sence de protéine pour la mutation (tableau 13.2).
Tableau 13.2. ![]()
![]() Corrélations entre le type de mutation, les conséquences cliniques et les risques génétiques.
Corrélations entre le type de mutation, les conséquences cliniques et les risques génétiques.
| Statut | Risque génétique |
Conséquences cliniques |
|
|---|---|---|---|
|
Chez l’homme |
Chez la femme |
||
| Normal | Expansions exceptionnelles | ||
| Intermédiaire | Expansions rares | ||
| Prémutation |
Expansion systématique, dans les 2 sexes Une femme prémutée peut transmettre une mutation complète |
Syndrome FXTAS : < 20 % à 60 ans, 75 % à 80 ans | Ménopause prématurée : risque cumulé 20 % |
| Mutation | Transmis uniquement par une femme mutée ou prémutée (jamais par un homme prémuté) | Déficience intellectuelle constante : syndrome de l’X fragile | Difficultés cognitives variables (50 % cas) |
Le diagnostic du syndrome de l’X fragile est difficile sur des bases cliniques, car les signes évocateurs sont discrets et peu spécifiques.
Il est donc souvent recherché en première intention dans l’exploration de toute déficience intellectuelle non syndromique.
Il n’y a pas de traitement curatif. La prise en charge est pluridisciplinaire : médicale (neuropédiatre, pédopsychiatre, généticien), médicosociale, éducative (scolarité adaptée) et rééducative (psychomotricité, orthophonie…).
2. X fragile chez les garçons porteurs d’une mutation complète
Chez l’homme, l’absence de protéine FRMP est systématiquement responsable d’une déficience intellectuelle, le syndrome de l’X fragile :
- déficience intellectuelle constante :
- retard de développement psychomoteur, en particulier sur le langage ;
- déficience intellectuelle modérée (QI habituellement entre 40 et 50) avec troubles des apprentissages ;
- troubles du comportement : hyperactivité, timidité avec fuite du regard ;
- troubles du spectre autistique (30 %) ;
- dysmorphie faciale (fig. 13.1) devenant plus évidente avec l’âge :
- visage allongé avec de grandes oreilles décollées ;
- macrocéphalie, front allongé, menton long et marqué ;
- signes inconstants : épilepsie, hyperlaxité ligamentaire, prolapsus de la valve mitrale… ;
- après la puberté : macro-orchidie.
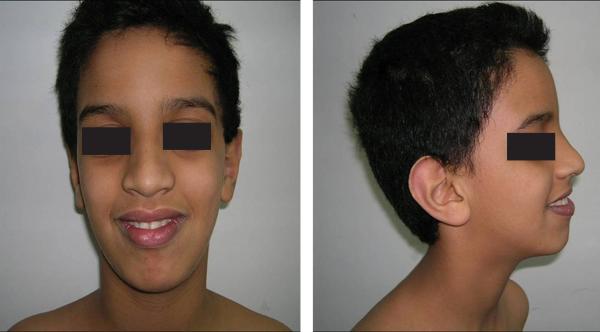
Fig. 13.1. ![]() Syndrome de l’X fragile : dysmorphie faciale.
Syndrome de l’X fragile : dysmorphie faciale.
3. X fragile chez les filles porteuses d’une mutation complète
Parmi les femmes porteuses d’une mutation complète, des difficultés cognitives de sévérité variable s’observent dans la moitié des cas, en relation avec l’inactivation physiologique plus ou moins biaisée d’un des deux chromosomes X.
Triade chez le garçon porteur d’une mutation complète = déficit intellectuel, dysmorphie faciale, macro-orchidie (après la puberté).
Déficit intellectuel si mutation complète = 100 % des garçons et 50 % des filles.
B. Conséquences cliniques d’une prémutation dans le gène FMR1
Le mécanisme pathogénique de la prémutation FRMP est un effet toxique des ARNm. Les personnes porteuses d’une prémutation n’ont pas de déficience intellectuelle mais peuvent développer des troubles neurologiques tardifs (Fragile X Tremor Ataxia Syndrome, FXTAS), plus commun chez les hommes, et, chez les femmes, une insuffisance ovarienne prématurée.
1. Prémutation chez les hommes
Le risque cumulé de développer un syndrome FXTAS est de l’ordre de 20 % entre 50 et 60 ans, de 75 % après 80 ans.
Cette maladie neurodégénérative progressive se traduit par des tremblements pseu-do-parkinsoniens, une ataxie cérébelleuse et un déclin cognitif avec troubles de la mémoire à court terme, dégradation des fonctions exécutives puis démence.
Il n’y a pas de traitement curatif.
2. Prémutation chez les femmes
La prévalence de l’insuffisance ovarienne prématurée est de 20 % (versus 1 % dans la population générale). Elle conduit à une ménopause précoce (aménorrhée > 4 mois avant 40 ans, avec ni-veau de FSH post-ménopausique), qui peut donc parfois être le signe révélateur du syndrome au sein d’une famille.
Prémutation : prédisposition à FXTAS et ménopause prématurée.
C. Examens paracliniques
La suspicion clinique doit être confirmée par un examen de biologie moléculaire. Pour rappel, les chromosomes X fragiles ne se voient pas sur un caryotype standard.
La recherche d’un X fragile doit être demandée spécifiquement au laboratoire.
Une PCR fluorescente permet d’amplifier l’ADN et d’évaluer le nombre de triplets CGG d’un allèle normal (exclusion du diagnostic) ou d’une prémutation courte.
Si la PCR ne détecte aucun allèle de petite taille chez le garçon ou un seul allèle chez une fille, le laboratoire recherche ensuite une grande expansion et établit le profil de méthylation par wes-tern blot.
Confirmation diagnostique = test moléculaire spécifique.
III. Conseil génétique et diagnostic prénatal
A. Conseil génétique
1 Généralités
Le conseil génétique est donné aux parents lors d’une consultation de génétique.
L’étude de l’arbre généalogique familial et des résultats de la génétique moléculaire permettent de déterminer le risque auquel les apparentés sont exposés.
2. Risque de transmission
Pour une femme porteuse d’une prémutation, la probabilité de transmettre une mutation com-plète dépend principalement de son nombre de triplets CGG : plus le nombre de triplets est grand, plus le risque de transmission est élevé.
Toutefois, si une femme prémutée a déjà engendré un enfant muté, la probabilité qu’elle trans-mette à nouveau une mutation plutôt qu’une prémutation est extrêmement élevée. En pratique, son risque d’avoir un garçon atteint est donc comparable à celui d’une femme mutée (50 %).
Pour une femme porteuse d’une mutation complète (ou une femme prémutée qui a déjà un en-fant porteur d’une mutation complète), la probabilité de transmettre une mutation complète à sa descendance est de 50 %.
Si elle attend un garçon, celui-ci aura un risque de 50 % d’hériter de la mutation complète et de présenter le syndrome de l’X fragile.
Si elle attend une fille, celle-ci a 50 % de risque d’hériter de la mutation complète, dont on sait qu’elle conduit à une déficience intellectuelle une fois sur deux.
Un homme porteur d’une prémutation transmettra cette prémutation à toutes ses filles.
Ses fils n’ont aucun risque.
Le dépistage des femmes conductrices est réalisé dans le cadre de la consultation de génétique. Le dépistage des hommes prémutés doit être proposé dans le cadre des consultations de diagnostic présymptomatiques (voir infra).
B. Diagnostic prénatal
Le diagnostic prénatal (DPN) doit être discuté avec toute femme porteuse d’une prémutation ou d’une mutation complète. L’X fragile entre dans les termes de la loi autorisant le DPN et l’interruption médicale de grossesse (IMG), en raison de la forte probabilité d’une affection d’une particulière gravité incurable au moment du diagnostic.
L’alternative au diagnostic prénatal est le diagnostic préimplantatoire. Cette technique a pour but d’identifier la présence de la mutation complète sur des embryons obtenus après fécondation in vitro. Le diagnostic moléculaire est réalisé sur une seule cellule de l’embryon prélevée au stade morula. Seuls sont transférés dans l’utérus des embryons portant un X non muté.
Conseil génétique car risque élevé de récurrence.
IV. Tests génétiques chez un mineur
A. Principes
Un test génétique ne peut être prescrit chez un mineur ou chez un majeur sous tutelle que si ce-lui-ci ou sa famille peuvent bénéficier de mesures préventives ou curatives immédiates.
Il n’est donc permis de tester un enfant non symptomatique que si un résultat anormal pourrait impacter sa prise en charge. Exceptionnellement, un mineur asymptomatique peut aussi être prélevé si son résultat est indispensable pour l’interprétation des données familiales.
B. Consentement éclairé et découvertes fortuites
L’information donnée avant de prescrire un test génétique doit notamment porter sur le cadre de l’examen (visée diagnostique, étude familiale, thérapeutique, conseil génétique…), les spécificités de la maladie recherchée, les limites des examens génétiques, le risque éventuel d’identification de caractéristiques génétiques sans relation directe avec la prescription et les obligations d’information de la parentèle.
Ces diagnostics fortuits sont fréquents lors de la réalisation d’examen chromosomique sur puce ADN et dans les tests fondés sur le séquençage de panels de gènes ou d’exomes.
C. Information de la parentèle
L’obligation d’information à la parentèle est applicable pour toute anomalie génétique grave dont les conséquences sont susceptibles de mesures de prévention, y compris de conseil génétique, ou de soins.
Sont concernées les situations impliquant :
- un risque de décès prématuré ;
- un risque de handicap sévère, le risque d’impossibilité d’autonomie à l’âge adulte ;
- une gravité particulière ou un caractère incurable au regard de l’état des connaissances et de la littérature au moment de la consultation.
Au moment de la restitution du résultat, le médecin doit dresser la liste des personnes à informer en se fondant sur l’arbre généalogique et le mode de transmission (apparentés du premier et du second degré, en général), et fournir au patient un document explicatif simple que ce dernier pourra diffuser à ses proches.
Si une personne refuse de transmettre elle-même l’information à ses proches à risque, elle peut autoriser par écrit le médecin à procéder à cette information. Elle est alors tenue de communiquer les coordonnées des intéressés dont elle dispose. Le non-respect de cette obligation est passible de poursuites pénales.
D. Diagnostic présymptomatique
Chez une personne asymptomatique mais ayant des antécédents familiaux, un test génétique ne peut être prescrit que dans le cadre d’une consultation médicale individuelle (art. R. 1131-5 du Code de la santé publique). Cette consultation est effectuée par un médecin œuvrant au sein d’une équipe pluridisciplinaire dotée d’un protocole type de prise en charge et déclarée auprès de l’Agence de la biomédecine.
Diffusion des résultats d’une anomalie génétique grave à la parentèle si mesures préventives possibles.