Objectifs pédagogiques
- Expliquer les bases du conseil génétique, les possibilités de dépistage et de diagnostic prénatal (modalités et options de prise en charge dans le cadre d’une maladie d’une particulière gravité).
- Expliquer la prescription des tests génétiques : organisation et aspects réglementaires (voir item 9).
- Expliquer les problèmes liés à la maladie et les retentissements de l’arrivée d’un enfant souffrant de maladie génétique sur le couple et la famille.
- Diagnostiquer la trisomie 21, en connaître l’évolution naturelle et les principales complications.
Avant de commencer…
La trisomie 21, ou syndrome de Down, est une maladie génétique fréquente, qui touche un fœtus sur 700. Elle est due le plus souvent à la présence d’un chromosome 21 surnuméraire (trisomie 21 libre) transmis dans 90 % des cas par la mère.
Le seul facteur de risque connu pour la survenue d’une trisomie 21 est l’âge maternel.
La confirmation diagnostique de la maladie repose sur un caryotype sanguin.
La trisomie 21 associe des anomalies développementales (dysmorphie faciale et malformations viscérales) à une déficience intellectuelle de sévérité variable. Elle est la première cause génétique de déficience intellectuelle.
De nombreuses complications somatiques peuvent survenir au cours du temps. Les malformations associées à la trisomie 21 sont potentiellement sévères et conditionnent le pronostic vital, alors que les difficultés neurodéveloppementales conditionnent le pronostic social.
Un accompagnement et une prise en charge multidisciplinaire médicale, rééducative et sociale sont indispensables à tous les âges de la vie pour la personne trisomique et sa famille.
L’espérance de vie est maintenant supérieure à 50 ans.
La trisomie 21 fait l’objet d’une proposition de dépistage systématique en cours de grossesse, fondé sur un prélèvement de sang maternel. La suspicion diagnostique peut être confirmée par un diagnostic pré-natal (DPN) invasif, suivi éventuellement d’une interruption médicale de grossesse (IMG).
Dépistage, diagnostic et IMG sont des choix personnels de la mère, nécessitant un consentement éclairé.
Du fait du diagnostic prénatal, l’incidence actuelle de la trisomie 21 est d’un cas pour 2 000 naissances.
I. Diagnostiquer une trisomie 21
A. Tableau clinique
Le tableau clinique ne comporte aucun signe pathognomonique. Seule une hypotonie marquée est quasi constante, mais non spécifique.
On retrouve à des degrés divers les éléments de la triade phénotypique de toute anomalie chromosomique : dysmorphie, malformations et retard psychomoteur.
1. Anomalies morphologiques
Le phénotype de la trisomie 21 est variable (tableau 12.1). Les malformations sont souvent identifiées avant la naissance. La dysmorphie, en particulier, résulte de l’accumulation chez un même individu de particularités morphologiques mineures et non spécifiques.
Reconnaître une trisomie 21 peut s’avérer difficile, en particulier dans un contexte ethnique non familier.
Tableau 12.1. ![]() Signes cliniques de la trisomie 21.
Signes cliniques de la trisomie 21.
| Dysmorphie craniofaciale (fig. 12.1) |
|
| Autres signes externes |
|
| Malformations cardiaques (50 %) |
|
| Malformations digestives |
|
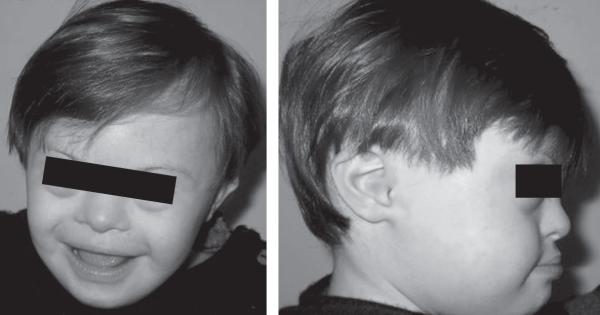
Fig. 12.1. ![]() Trisomie 21 : dysmorphie craniofaciale.
Trisomie 21 : dysmorphie craniofaciale.
2. Retard psychomoteur et manifestations psychiatriques
À l’hypotonie néonatale quasi constante succède un retard des acquisitions psychomotrices, avec un retard de langage marqué.
Le déficit cognitif est très variable. Si le QI moyen se situe autour de 45, il peut varier entre 25 (déficience intellectuelle sévère) et 80 (niveau normal faible). La lecture et l’écriture sont possibles pour deux tiers des sujets.
La trisomie 21 est un facteur de risque pour l’autisme, qui s’observe dans 3 % des cas.
Avec l’âge, les patients sont à risque de développer une démence de type Alhzeimer avec une régression (> 50 % des cas).
3. Autres complications
De nombreuses complications peuvent survenir, notamment des maladies auto-immunes (tableau 12.2).
Les sujets trisomiques 21 ont des difficultés à décrire leurs symptômes ou à localiser et quantifier leurs douleurs. Le diagnostic des complications peut donc être retardé ou négligé et les bilans systématiques sont importants. Une attention particulière doit être prêtée aux changements de l’humeur, aux modifications inattendues de poids…, qui peuvent signer une complication secondaire.
Tableau 12.2. ![]() Complications de la trisomie 21.
Complications de la trisomie 21.
| Pathologie | Incidence* | Remarque |
|---|---|---|
| Obésité | 30–50 % | Aggravée par la sédentarité, l’absence d’activité physique et la boulimie |
| Diabète de type 1 | 1 % | |
| Diabète de type 2 | Secondaire à l’obésité Dépistage systématique à l’âge adulte | |
| Hypothyroïdie et thyroïdite | 4–18 % | Dépistage annuel |
| Maladie cœliaque | 5 % | Dépistage clinique |
| Syndrome d’apnées obstructives du sommeil (SAOS) | > 50 % | Dépistage systématique à 4 ans par polysomnographie |
| Infections ORL récurrentes | 60 % | Déficit non spécifique de l’immunité humorale et cellulaire |
| Surdité | 45–75 % | 2 % de surdité de perception congénitale Examen de dépistage annuel |
| Strabisme et myopie | 60 % | |
| Cataracte | 15 % | Peut se manifester à tous les âges Examen systématique annuel (tous les 3 ans chez l’adulte) |
| Épilepsie | 1–10 % | Risque de syndrome des spasmes épileptiques infantiles |
| Polycaries et maladie parodontale | 90 % | Soins sous anesthésie générale |
| Hyperleucocytose du nourrisson | 4–10 % | |
| Leucémie aiguë myéloblastique | 1 % | |
| Instabilité atlanto-axoïdienne | 2 % | Pas de dépistage radiologique Prudence en cas d’intubation |
| Ostéoporose | Chez l’adulte (surtout femme ménopausée) | |
| Maladie d’Alzheimer | 50 % | Après 40 ans |
| Stérilité | Chez les hommes (les femmes ont une fertilité normale) |
* Les chiffres d’incidence sont donnés à titre indicatif.
Triade d’anomalies chromosomiques = dysmorphie, malformations et retard psychomoteur.
Complications principales : obésité, infections ORL et SAOS, dysthyroïdie, épilepsie.
B. Diagnostic génétique
1. Investigations cytogénétiques
La confirmation diagnostique de la trisomie 21 requiert un caryotype sanguin, même si le diagnos-tic clinique paraît évident. Elle nécessite le consentement écrit des parents.
Les quatre formes cytogénétiques de trisomie 21 sont résumées dans le tableau 12.3.
Tableau 12.3. ![]() Formes cytogénétiques de trisomie 21.
Formes cytogénétiques de trisomie 21.
| Trisomie 21 libe homogène | 95 % | 47 chromosomes (47,XX,+ 21 ou 47,XY,+ 21) Caryotype parental inutile |
| Trisomie 21 libre en mosaïque | 2 % | Des cellules à 47 chromosomes dont 3 chromosomes 21 coexistent avec des cellules à 46 chromosomes dont 2 chromosomes 21 Caryotype parental inutile |
| Trisomie 21 par translocation roberstonienne | 3 % | 46 chromosomes dont un chromosome recombinant Caryotype parental indispensable : 50 % de ces translocations sont héritées d’un parent à 45 chromosomes, porteur d’une translocation équilibrée robertsonienne impliquant les chromosomes acrocentriques (13, 14, 15, 21, 22) |
| Trisomie 21 par translocation réciproque | Rare | 46 chromosomes dont un chromosome recombinant Caryotype parental indispensable (50 % de formes héritées) |
2. Bases du conseil génétique
Le conseil génétique s’adresse aux couples ayant un risque accru d’avoir un fœtus atteint de trisomie 21 et aux couples ayant eu un fœtus ou un enfant atteint de trisomie 21 ou des antécédents familiaux de trisomie 21. Il a pour but d’évaluer le risque de trisomie 21 pour un futur enfant de ce couple et d’envisager les stratégies de prévention.
Le risque a priori de trisomie 21 est fonction de l’âge maternel : 1 pour 1 500 naissances à 20 ans, 1 pour 1 000 à 30 ans, 1 pour 350 à 35 ans, 1 pour 100 à 40 ans, 1 pour 30 à 45 ans.
Le risque pour une femme trisomique 21 d’avoir un enfant trisomique est de 1/3.
Une trisomie 21 libre est due à une non-disjonction méiotique accidentelle. L’examen des caryotypes parentaux est inutile.
Pour un couple ayant eu un fœtus ou un enfant atteint d’une trisomie 21 libre, le risque de récurrence pour un nouvel enfant est de 1 %. Ceci s’explique par l’existence de mosaïques germinales parentales indétectables.
Dans le cas d’une trisomie 21 par translocation, le caryotype des parents est indispensable.
Si les caryotypes des parents sont tous les deux normaux (translocation de novo), le risque de récurrence est de 1 %. Si l’un des parents a une translocation équilibrée, le risque de récurrence varie de 10 % à 100 % (dans le cas exceptionnel d’un parent porteur d’une translocation robertsonienne impliquant ses deux chromosomes 21). Il est plus élevé si c’est la mère qui porte la translocation.
Lorsqu’un parent porte une translocation équilibrée, les apparentés majeurs de cette personne doivent être informés de leur risque d’être porteurs (voir la section sur l’information de la parentèle, dans le chapitre 13 consacré à l’X fragile). Un caryotype doit leur être proposé.
Confirmation = caryotype sanguin : trisomie 21 libre, complète et homogène (95 % des cas).
Risque de récurrence faible pour une trisomie 21 libre : 1 %.
II. Prendre en charge et assurer le suivi d’un enfant trisomique
A. Annonce diagnostique
Toute annonce d’une pathologie grave doit se faire dans un endroit calme pour une écoute et une disponibilité optimale, si possible en présence des deux parents.
Une information adaptée et loyale doit être délivrée aux deux parents sur la maladie, son évolution naturelle et ses complications, sa prise en charge médicale et sociale.
L’annonce d’un diagnostic de trisomie 21 est toujours délicate, car c’est une affection que « tout le monde connaît », ce qui signifie beaucoup de préjugés et de figurations erronées. Il est inutile d’inonder les parents d’informations médicales complexes à ce stade et il faut éviter les affirmations péremptoires sur le pronostic ou le devenir : l’avenir d’un enfant atteint de trisomie 21 est aussi imprévisible que celui de tout enfant. Une présentation positive de la prise en charge est in-dispensable. Il faut informer les parents de l’existence de consultations spécialisées dans le suivi des enfants trisomiques et de l’existence des associations de parents.
Ne pas hésiter à revoir rapidement les parents : il est plus raisonnable de focaliser le premier entretien sur les questions les plus urgentes et d’approfondir la discussion lors d’entretiens ultérieurs, car la sidération qui accompagne souvent ce diagnostic inattendu ne permet pas aux parents d’appréhender toutes les implications ni de formuler toutes les questions.
En période anténatale, l’IMG doit être envisagée et discutée au cours de la consultation.
Il faut tenir compte de la culture et des positions éthiques, philosophiques ou des convictions religieuses des parents. L’objectif est de fournir des informations claires et précises, puis d’accompagner les parents dans le choix qu’ils ont fait, en conservant une neutralité bienveillante, quelle que soit leur option.
B. Prise en charge et suivi
1. Prise en charge médicale
La prise en charge médicale est pluridisciplinaire et à vie. Elle associe une surveillance clinique, biologique et morphologique, et la détection systématique des complications tardives.
Une surveillance pédiatrique régulière est indispensable : croissance, alimentation, développe-ment psychomoteur, dépistage des anomalies sensorielles… (tableau 12.2).
Les examens paracliniques au diagnostic comprennent un bilan hématologique (NFS), un bilan thyroïdien, une échographie cardiaque et rénale.
La recherche de surdité est indispensable dès la première année, car une déficience auditive va contribuer aux difficultés d’apprentissage du langage.
L’examen ophtalmologique régulier est indispensable (réfraction, strabisme, cataracte).
Les dysthyroïdies doivent être recherchées annuellement ; la possibilité de maladie cœliaque doit rester à l’esprit.
Le dépistage du SAOS par polysomnographie est recommandé à 4 ans.
2. Prise en charge sociale et paramédicale
Les mesures sociales et éducatives sont détaillées dans le chapitre 53 « Handicap ».
Dès l’âge de 3 à 6 mois, une rééducation en psychomotricité peut être prescrite dans le cadre d’une prise en charge dans un CAMSP (jusqu’à l’âge de 6 ans), un SESSAD, ou par un praticien libéral formé. La rééducation orthophonique peut être ajoutée dès l’âge de 1 an et doit être instaurée à l’âge de 3 ans.
Un suivi par un psychologue peut être utile, surtout après l’entrée en CP et à la puberté.
La scolarité est adaptée au niveau intellectuel : enseignement habituel avec une AESH, ULIS ou IME.
Annonce diagnostique avec tact et empathie.
Prise en charge médicale multidisciplinaire à vie.
C. Suivi de l’adulte trisomique
Un dépistage de l’hypothyroïdie, du diabète, de la maladie cœliaque et de la cataracte sont indispensables tous les 2 ou 3 ans.
Les dépistages systématiques du cancer du côlon, du sein, du col de l’utérus doivent être pratiqués.
Les troubles de l’humeur, des difficultés comportementales, un repli doivent faire rechercher une cause organique (douleurs orthopédiques, œsophagite, maladie d’Alzheimer…) avant toute prise en charge symptomatique.
L’entretien des acquis après la période scolaire est fondamental pour maintenir le niveau d’autonomie et d’intégration sociale atteint.
Beaucoup de patients trisomiques peuvent accéder au marché du travail comme travailleur handicapé ou dans des structures adaptées (ESAT).
Les personnes trisomiques ont pour la plupart la capacité de mener une vie affective normale et peuvent être sexuellement actives. Une contraception adaptée doit être discutée pour les adolescentes et femmes trisomiques.
D. Accompagnement familial
La naissance d’un enfant atteint de trisomie 21 est source d’un bouleversement au sein des fa-milles. Elle peut conduire à un rejet affectif de l’enfant atteint (parfois à un abandon ou de la mal-traitance), à une surprotection (au détriment de l’accompagnement affectif de la fratrie), à des tensions parentales (dépression, divorce), à un déni du handicap à venir.
L’accompagnement d’un enfant trisomique et de sa famille doit être assuré dès le diagnostic (anténatal ou postnatal), ainsi qu’à l’occasion du conseil génétique. L’aide des associations de parents peut être utile pour ces familles.
La guidance parentale est indispensable tout au long de l’enfance, pour investir les potentialités des enfants sans nier le handicap, pour accompagner les familles dans l’autonomisation de leur enfant. Il est important d’impliquer l’adolescent dans l’élaboration de son projet de vie et dans l’éducation à l’autodétermination et la socialisation
III. Dépistage et diagnostic prénataux de la trisomie 21
A. Principes généraux
La surveillance systématique d’une grossesse comprend l’évaluation du risque fœtal de trisomie 21, qui repose sur l’âge maternel et sur la mesure échographique de la clarté de la nuque au premier trimestre, le dosage de marqueurs sériques maternels du premier trimestre et la réalisation de trois échographies, à 12, 22 et 32 SA.
Ce dépistage prénatal doit être systématiquement proposé au cours de la grossesse.
Il a pour but d’identifier des femmes qui ont un risque accru de porter un fœtus avec trisomie 21.
Seul un caryotype fœtal obtenu par un diagnostic prénatal (DPN) invasif permet toutefois de poser un diagnostic de certitude.
Si le dépistage indique qu’une femme a un risque de trisomie 21 supérieur au seuil admis, un DPN est proposé. En cas de positivité, le DPN peut amener à l’interruption de la grossesse.
Chaque étape de ce processus est soumise à consentement écrit.
Une mère qui ne souhaite aucun examen permettant d’évaluer « le risque que le fœtus présente une affection susceptible de modifier le déroulement ou le suivi de la grossesse ou des examens à visée de diagnostic doit signer un document précisant son refus » (arrêté du 14 janvier 2014).
Par ailleurs, le dépistage sur marqueurs sériques, l’échographie de dépistage, l’échographie de diagnostic et les examens invasifs sont soumis à des consentements écrits distincts.
B. Dépistage
1. Évaluation du risque de trisomie 21 fœtale : approche classique
Échographies de suivi de grossesse
L’échographie prénatale poursuit deux buts : suivre le déroulement de la grossesse (échographie obstétricale) et rechercher des anomalies du développement fœtal (échographie morphologique).
L’échographie du premier trimestre (dite de datation) permet la mesure de la clarté de la nuque, normalement inférieure à 3 mm.
Elle doit être effectuée entre le début de la 11e SA et la fin de la 13e SA par un échographiste identifié au sein d’un réseau de périnatalité.
Un hygroma kystique de la nuque (hygroma colli) est fréquemment associé à une anomalie chromosomique fœtale.
L’échographie de 22 SA permet de rechercher une anomalie malformative évocatrice.
L’échographie de 32 SA peut identifier un RCIU tardif.
Calcul combiné de risque
Des algorithmes permettent de moduler le risque a priori de trisomie 21, déterminé par l’âge maternel, en intégrant dans un « calcul combiné » la mesure de la clarté nucale au premier tri-mestre, les marqueurs du premier trimestre et des éléments correctifs : le poids, le tabagisme, l’origine géographique (européenne/nord-africaine, afro-caribéenne, asiatique), la gémellité…
Le seuil de positivité a été fixé à 1/250 (risque a priori d’une femme de 37 ans).
2. Évaluation du risque de trisomie 21 fœtale par étude de l’ADN libre circulant
Le plasma contient de l’ADN libre circulant (ALC), constitué de très petits fragments, provenant principalement de la dégradation des cellules endothéliales. Durant la grossesse, dès la 7e semaine, la destruction des cellules placentaires fœtales au contact de la circulation maternelle constitue une deuxième source d’ALC (5 à 10 % du total). Sa demi-vie ne dépasse pas 2 jours.
Ces techniques permettent d’établir en quelques jours, en un seul temps analytique, la séquence individuelle de plusieurs dizaines de millions de fragments d’ADN isolés du sang maternel, afin d’en déterminer l’origine chromosomique. Si le fœtus est trisomique, la technique identifie un excès relatif significatif de fragments d’ADN issus du chromosome 21 comparativement aux autres (sensibilité et spécificité > 99 %).
En 2017, la HAS recommande de proposer le dépistage par NIPT (Non-Invasive Prenatal Testing) de la trisomie 21 aux femmes dont le niveau de risque estimé est compris entre 1 sur 1 000 et 1 sur 51 par le dépistage classique. Tout dépistage par NIPT anormal doit être confirmé par caryotype fœtal qui seul permet de poser un diagnostic de certitude. Pour les femmes dont le risque est supérieur ou égal à 1 sur 50, la réalisation d’un caryotype fœtal d’emblée est maintenue mais en intégrant la possibilité pour celles qui le souhaiteraient de réaliser dans un premier temps un test ADN.
3. Résultats du dépistage de la trisomie 21
La dimension éthique de cette procédure de diagnostic anténatal ne doit pas être sous-estimée car plus de 95 % des diagnostics anténatals de trisomie 21 conduisent à une IMG.
Les enjeux doivent être expliqués de façon précise aux parents pour que leur décision soit prise en connaissance de cause ; ce qui reste de toute façon très difficile.
Dépistage prénatal = clarté de nuque et autres anomalies échographiques, marqueurs sériques maternels. Calcul combiné de risque. Accompagnement des familles.
C. Diagnostic prénatal
1. Indications
Le caryotype fœtal permet un diagnostic anténatal de trisomie 21.
Ses indications répondent au terrain maternel et familial, ainsi qu’aux arguments de suspicion diagnostique anténatals :
- remaniement chromosomique parental ;
- antécédent d’un fœtus ou d’un enfant porteur d’une anomalie chromosomique ;
- risque calculé ≥ 1/50, dépistage par NIPT anormal ;
- signes d’appel échographiques.
2. Modalités
Deux méthodes de prélèvement sont possibles : la choriocentèse ou biopsie de trophoblaste (BT) pour étude des villosités choriales, et l’amniocentèse ou ponction de liquide amniotique (PLA). La cordocentèse (ou ponction de sang fœtal) est réservée à des situations très particulières nécessitant un dosage plasmatique fœtal.
Le recueil du consentement éclairé écrit de la mère est indispensable. Elle doit être informée de l’objectif de l’examen, des procédures, de la possibilité de dépistage d’autres anomalies chromosomiques que la trisomie 21 et des risques de fausse couche iatrogène (1 % pour la BT et 0,5 % pour la PLA).
Lorsqu’une anomalie précise est recherchée, une hybridation in situ à l’aide de sondes fluorescentes (technique FISH) permet un marquage en interphase du noyau de cellules obtenues par amniocentèse, sans culture préalable.
En cas de DPN pour anomalies morphologiques fœtales, un caryotype moléculaire par analyse chromosomique sur puce ADN (ACPA) peut être proposé, afin d’identifier des remaniements chromosomiques de petite taille ou pour mieux définir une anomalie identifiée sur un caryotype fœtal standard.
D. Interruption médicale de grossesse
Un diagnostic prénatal de trisomie 21 peut conduire les parents à faire la demande d’une inter-ruption médicale de grossesse (IMG), qui sera évaluée au sein d’un centre pluridisciplinaire de diagnostic prénatal (CPDPN).
L’aide d’un psychologue est systématiquement proposée à la mère ou au couple.
Diagnostic prénatal = caryotype fœtal avec accord écrit maternel.
Dimension éthique du recours éventuel à une interruption médicale de grossesse.
Références

|
HAS. Protocole national de diagnostic et de soins (PNDS) – Trisomie 21. 2020. https://www.has-sante.fr/upload/docs/application/pdf/2020-01/pnds_trisomie_21.pdf |

|
Bull MJ. Committee on Genetics. Health supervision for children with Down syndrome. Pediatrics 2011;128:393–406. http://pediatrics.aappublications.org/content/128/2/393 |