Items, objectifs pédagogiques
Item 215 – Purpuras chez l’adulte et l’enfant
- Argumenter les principales hypothèses diagnostiques et justifier les examens complé-mentaires pertinents.
Item 216 – Syndrome hémorragique d’origine hématologique
- Diagnostiquer un syndrome hémorragique d’origine hématologique.
- Interpréter les examens courants d’hémostase.
Item 329 – Transfusion sanguine et produits dérivés du sang : indications, complications. Hémovigilance
- Expliquer les risques transfusionnels, les règles de prévention, les principes de traçabi-lité et d’hémovigilance.
- Prescrire une transfusion des médicaments dérivés du sang.
Avant de commencer…
Le purpura est une tache hémorragique due à l’extravasation de sang dans le derme.
Cette lésion est liée à une anomalie de l’hémostase primaire, impliquant les vaisseaux ou les plaquettes.
La démarche sémiologique doit apprécier avant tout l’existence d’un contexte infectieux. Tout purpura fébrile doit faire évoquer le diagnostic de purpura fulminans (purpura vasculaire, urgence vitale). La grande majorité des purpuras fébriles est cependant reliée à des causes virales non sévères.
L’évaluation de la gravité d’un syndrome hémorragique ou la recherche d’arguments en faveur d’une hémopathie maligne sont également essentielles.
L’examen indispensable d’orientation diagnostique est l’analyse quantitative et qualitative de la NFS (frottis). Elle permet de guider l’enquête étiologique : purpuras non thrombopéniques et purpuras thrombopéniques, et dans ce dernier cas thrombopénie isolée ou non.
Seule la démarche diagnostique en cas de purpura « aigu » est traitée.
Les « spécificités pédiatriques » seront détaillées, avec les points clés de deux causes fréquentes chez l’enfant : le purpura rhumatoïde (purpura vasculaire) et le purpura thrombopénique immunologique (purpura thrombo-pénique). Le purpura fulminans est traité dans le chapitre « Choc septique » (voir chapitre 67).
I. Diagnostiquer un purpura et planifier la prise en charge
A. Identifier un purpura
Le diagnostic de purpura est exclusivement clinique.
Il s’agit d’une lésion cutanée et/ou muqueuse hémorragique, de couleur rouge ou pourpre, ne s’effaçant pas à la vitropression, signant une extravasation de globules rouges à l’extérieur des vaisseaux.
Ne pas le méconnaître impose de déshabiller complètement l’enfant lors de l’examen clinique (en enlevant les chaussettes et la couche chez un nourrisson) et de regarder en intrabuccal.
L’analyse sémiologique du purpura est essentielle (tableau 24.1) :
- inspection : pétéchies, ecchymose(s), nécrose(s) ;
- palpation : maculeux (non palpable), infiltré (palpable) ;
- topographie : localisé (visage, zones de frottements ou traumatismes, régions déclives, membres inférieurs ou lombes, fesses, extrémités distales, muqueuses) ou disséminé ;
- autres signes hémorragiques extériorisés ou non.
Tableau 24.1. ![]() Comparaison des purpuras thrombopénique et vasculaire
Comparaison des purpuras thrombopénique et vasculaire
| Purpura thrombopénique | Purpura vasculaire |
|---|---|
| Voir infra fig. 24.3 | Voir infra fig. 24.4 |
| Caractère maculeux Pas de prédominance déclive ± Atteinte muqueuse ± Ecchymose(s) |
Caractère infiltré Prédominance déclive Jamais d’atteinte muqueuse Polymorphisme lésionnel |
Le purpura pétéchial est constitué d’éléments punctiformes rouge pourpre.
Le purpura ecchymotique est constitué de nappes bleu violacé.
Les lésions purpuriques distales peuvent évoluer vers un caractère nécrotique et parfois ulcéré.
Tache rouge ne s’effaçant pas à la vitropression = purpura.
Entourer au stylo les lésions purpuriques afin d’apprécier leur évolution.
B. Apprécier la gravité
Sepsis grave lié à une possible infection invasive à méningocoque :
- caractéristiques sémiologiques de purpura fulminans (fig. 24.1) :
- caractère rapidement extensif ;
- ≥ 1 élément nécrotique ou ecchymotique de diamètre ≥ 3 mm ;
- fièvre élevée, frissons, extrémités froides, marbrures, TRC ≥ 3 s, tachycardie, hypotension ;
- atteinte des extrémités.
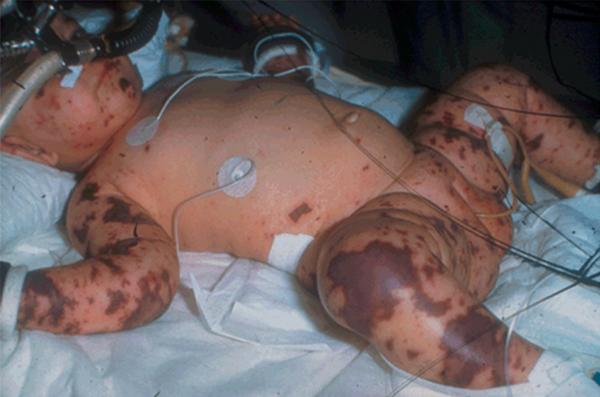
Fig. 24.1. ![]() Purpura fulminans.
Purpura fulminans.
Syndrome hémorragique lié à une thrombopénie profonde :
- saignement extériorisé, hématurie macroscopique, ménométrorragies ;
- atteinte des muqueuses (bulles hémorragiques intrabuccales, gingivorragies, épistaxis) ;
- signes extracutanés évoquant un saignement viscéral :
- céphalées, signes méningés, troubles de conscience (hémorragie intracrânienne) ;
- syndrome abdominal aigu (hémorragie digestive/gynécologique/urologique, rupture de rate) ;
- collapsus, syndrome anémique lié à une hémorragie aiguë.
Signes reliés à une hémopathie :
- atteinte des autres lignées médullaires :
- anémie : syndrome anémique grave ;
- neutropénie : fièvre, angine, stomatite ;
- syndrome tumoral : adénopathies, hépatosplénomégalie, atteinte testiculaire, douleurs osseuses ;
- altération de l’état général récente.
Signes reliés à une autre cause potentiellement grave, telle une microangiopathie thrombotique (MAT, SHU) :
- atteinte rénale, HTA ;
- diarrhée récente (volontiers glairosanglante).
C. Connaître les mesures d’urgence
1. Purpura fébrile
Mesures systématiques :
- identifier des signes évocateurs de purpura infectieux sévère ;
- surveiller de manière rapprochée l’enfant :
- scope cardiorespiratoire, examens cliniques répétés ;
- entourer les lésions et surveiller leur extension.
En cas de signes évocateurs d’un purpura fulminans :
- injection IM/IV de C3G au mieux après une hémoculture ;
- pose de deux VVP, voire mise en place d’une voie intra-osseuse ;
- expansion volémique avec un soluté cristalloïde isotonique balancé (à défaut, possibilité d’utiliser un soluté cristalloïde non balancé = NaCl 0,9 %) : prescrire un ou plusieurs bolus de 10 ml/kg (maximum 500 ml par remplissage), chacun administré le plus rapidement possible sur 10 à 15 min, jusqu’à un volume total de 40 à 60 ml/kg dans la 1re heure de prise en charge (voir chapitre 67) ; réévaluation hémodynamique très régulière ;
- transfert médicalisé en réanimation.
2. Purpura avec syndrome hémorragique
Mesures systématiques :
- identifier des signes hémorragiques majeurs ;
- surveiller de manière rapprochée l’enfant :
- scope cardiorespiratoire, examens cliniques répétés ;
- syndrome anémique.
En cas de signes évocateurs d’un syndrome hémorragique sévère :
- arrêt d’un saignement actif : compression ;
- pose de deux VVP, bilan prétransfusionnel : groupe (deux déterminations), Rhésus, RAI ;
- ± transfusion de produits sanguins labiles (CGR, plaquettes).
La décision de transfusion de plaquettes est discutée selon l’étiologie.
Elle peut être indiquée en cas de thrombopénie d’origine centrale. Elle n’est pas indiquée en cas de thrombopénie d’origine périphérique, sauf en cas de complication hémorragique grave.
3. Purpura avec symptômes évocateurs d’une cause centrale
Mesures systématiques :
- rechercher des signes de gravité symptomatique ;
- prise en charge de l’atteinte des autres lignées sanguines.
Programmer un myélogramme en milieu spécialisé.
Gravité : syndrome hémorragique, sepsis, purpura rapidement extensif, syndrome tumoral.
Suspicion de purpura fulminans : injection IM/IV de C3G et prise en charge du sepsis.
D. Conduire l’enquête étiologique
1. Enquête clinique minutieuse
Anamnèse :
- antécédents familiaux :
- thrombopénie ou thrombopathie constitutionnelle ;
- pathologie dysimmunitaire (lupus, thyroïdite…).
- terrain :
- âge, sexe, origine ethnique ;
- épisodes antérieurs de signes hémorragiques (interventions, traumatismes), NFS antérieures ;
- existence d’un contexte particulier :
- infection virale, contage infectieux, séjour à l’étranger, prise médicamenteuse ;
- statut vaccinal (pneumocoque, méningocoque), vaccin récent (ROR) ;
- signes cliniques :
- fièvre ;
- hémorragie extériorisée (épistaxis…) ou non (céphalées, douleurs abdominales) ;
- altération de l’état général, douleurs osseuses ;
- arthralgies, vomissements, toux, dysnée, autre éruption cutanée.
Examen physique :
- signes de sévérité symptomatique et de cause potentiellement grave ;
- signes orientant vers une étiologie : infection en cours, syndrome tumoral, diarrhée, HTA, anomalies articulaires, orchite, palpation abdominale douloureuse.
2. Enquête paraclinique
NFS-plaquettes avec réticulocytes et frottis sanguin
Ce sont les examens indispensables pour l’orientation diagnostique.
La NFS permet de distinguer : purpuras thrombopéniques (plaquettes < 150 G/l) et non thrombopé-niques.
La concertation avec le biologiste permet de décider des examens spécialisés ultérieurs, en fonction du frottis sanguin qui peut apporter des informations précieuses au niveau de l’ensemble des lignées :
- agrégats plaquettaires, taille et aspect des plaquettes ;
- syndrome mononucléosique, cellules malignes, corps de Döhle… ;
- schizocytes, réticulocytose.
En cas de purpura fébrile avec des signes cutanés et/ou hémodynamiques évoquant un purpura fulmi-nans, aucun examen n’est nécessaire avant l’injection urgente d’une antibiothérapie probabiliste (voir chapitre 67).
Examens de première intention
Systématiques :
- NFS-plaquettes, réticulocytes, frottis sanguin (vérification de l’absence de cellules anormales, de schi-zocytes ou d’aggrégats plaquettaires) ;
- TP-TCA, fibrinogène (recherche d’une CIVD) ;
- créatininémie, BU (recherche d’hématurie et de protéinurie).
Selon contexte :
- groupe, Rhésus, RAI : en cas de syndrome hémorragique ;
- test de Coombs érythrocytaire, haptoglobine et bilirubine : en cas d’anémie et/ou taux de réticulo-cytes élevé en faveur d’une hémolyse ;
- scanner cérébral et fond d’œil : en cas de céphalées et/ou signes neurologiques anormaux ;
- imagerie abdominale : en cas de syndrome abdominal aigu (vomissements, subocclusion, méléna, ménométrorragies, traumatisme) ;
- bilan infectieux (hémoculture, CRP, ± autres examens selon contexte) : en cas de fièvre ; recherche de paludisme et dengue : en cas de séjour récent en zone endémique.
Examens de seconde intention selon le contexte
- Myélogramme : en cas de syndrome tumoral ou d’anomalies des autres lignées sanguines.
- Bilan d’hémostase primaire : en cas d’histoire hémorragique personnelle et/ou familiale chronique dosage de facteur Willebrand (antigène et activité), tests d’agrégation plaquettaire et d’expression des glycoprotéines plaquettaires (recherche d’une thrombopathie).
- Sérologies VIH, hépatites B et C.
- VS, bilan de vascularite/auto-immun : facteurs antinucléaires (FAN), anticorps anti-ADN, ANCA ; re-cherche d’atteinte d’organe associée.
- Dosage pondéral des immunoglobulines, phénotypage lymphocytaire.
Rechercher : fièvre, thrombopénie constitutionnelle familiale, syn-drome anémique.
Examens indispensables en cas de purpura : NFS-plaquettes avec réticulocytes et frottis sanguin.
3. Principales causes de purpura chez l’enfant
La conduite à tenir pratique face à des lésions purpuriques chez un enfant est synthétisée dans la fi-gure 24.2.
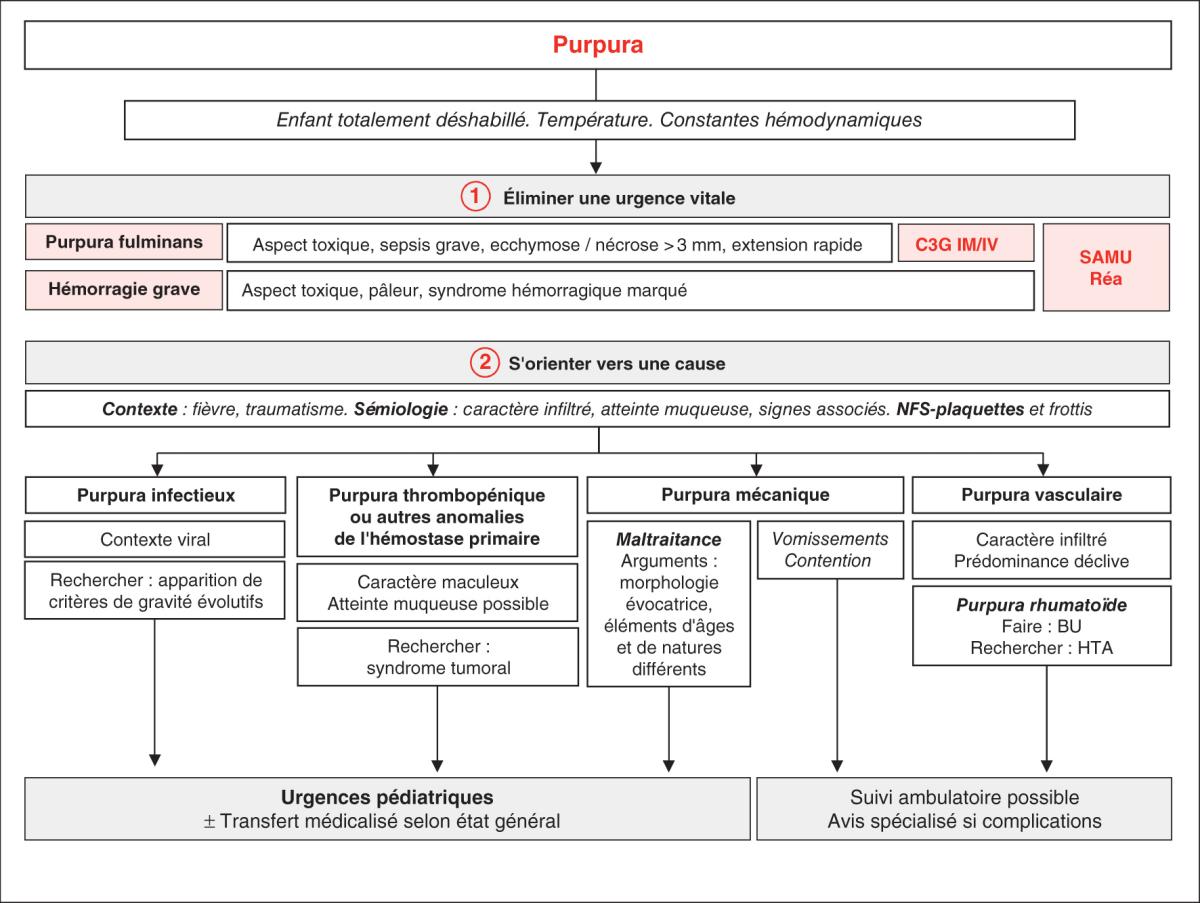
Fig. 24.2. ![]() Conduite diagnostique en cas de purpura chez l’enfant.
Conduite diagnostique en cas de purpura chez l’enfant.
D’après : Benoist G. Algorithme de prise en charge du purpura. In : Bourrillon A., Benoist G., Chabrol B., Ché-ron G., Grimprel E. Pédiatrie pour le praticien. 7e édition. 2020. © Elsevier Masson SAS. Tous droits réservés.
- En cas de purpura fébrile, il faut redouter en premier lieu un purpura fulminans (exceptionnel mais grave, voir chapitre 67) ; la plupart du temps, il s’agit d’un purpura infectieux non sévère.
- En cas de purpura non thrombopénique, la cause la plus fréquente est le purpura rhumatoïde (pur-pura vasculaire, voir infra) et les causes mécaniques (pétéchies liées au point de compression d’un garrot, pétéchies du territoire cave supérieur liées à la toux ou aux vomissements).
- En cas de purpura thrombopénique, la cause hématologique la plus fréquente est le purpura throm-bopénique immunologique, mais une cause centrale (hémopathie) doit toujours être écartée.
Les autres principales causes de purpura sont précisées dans le tableau 24.2.
Tableau 24.2. ![]()
![]() Principales causes de purpura.
Principales causes de purpura.
| Non thrombopéniques | Thrombopéniques (PT) | ||
|---|---|---|---|
| Purpura vasculaires | Thrombopathies | PT périphériques | PT centraux |
|
|
|
|
E. Savoir prescrire une transfusion de plaquettes
Une transfusion de plaquettes n’est pas indiquée en cas de purpura avec thrombopénie relié à un PTI, sauf en cas d’hémorragie sévère ou neurologique, intra-abdominale, extériorisée abondante ou urgence chirurgicale à risque.
On retient habituellement comme seuil transfusionnel une thrombopénie < 20 G/l chez un enfant traité par chimiothérapie pour une leucémie aiguë ou encore < 50 G/l chez un enfant ayant une tumeur cérébrale ou d’autres situations telles qu’un traitement anticoagulant, un saignement actif (en particulier au cours d’une CIVD).
Avant la transfusion :
- connaître les antécédents de transfusion de l’enfant (nombre, accidents éventuels) ;
- information des parents ± de l’enfant sur le rapport bénéfice/risque, accord parental ;
- bilan prétransfusionnel (± éventuels prélèvements à visée étiologique).
Commande des concentrés plaquettaires d’aphérèse (CPA) ou mélanges de concentrés plaquettaires (MCP) :
- nom, prénom, âge, poids de l’enfant, chiffre de plaquettes, tolérance clinique ;
- date, identification du prescripteur, signature, degré d’urgence ;
- quantité à transfuser = 1 unité plaquettaire pour 5 à 9 kg de poids (maximum 9 unités).
Modalités de la transfusion :
- en débit libre ;
- surveillance rapprochée des paramètres vitaux (température, hémodynamique, conscience) ;
- attention au volume total chez le petit enfant.
Après la transfusion : - traçabilité : notification dans les dossiers transfusionnel et médical et le carnet de santé ;
- suivi de l’efficacité de la transfusion : évolution clinique, ± NFS de contrôle.
Transfusion : CPA ou MCP, débit libre, surveillance clinique, NFS.
II. Points clés à propos de deux causes de purpura
A. Purpura rhumatoïde
1. Généralités
C’est la vascularite primitive la plus fréquente chez l’enfant. Il s’agit d’une vascularite leucocytoclasique touchant les petits vaisseaux par dépôts de complexes immuns circulants fixant majoritairement des IgA dans la paroi des capillaires de la peau, du tube digestif, ainsi que dans le mésangium des glomérules rénaux.
La physiopathologie de la maladie n’est pas bien connue, mais des facteurs immunologiques, génétiques et environnementaux semblent jouer un rôle. Des facteurs déclenchants ont parfois été rapportés : rhinopharyngite, vaccination récente, médicament.
Cette pathologie concerne plus fréquemment le garçon avec un pic d’incidence entre 4 et 6 ans, en période automno-hivernale.
2. Diagnostic
Enquête clinique
Le diagnostic de purpura rhumatoïde est avant tout clinique et repose sur une triade (tableau 24.3) : purpura (fig. 24.3), manifestations articulaires, douleurs abdominales.
Tableau 24.3. ![]() Triade clinique du purpura rhumatoïde.
Triade clinique du purpura rhumatoïde.
| Purpura |
|
| Manifestations articulaires |
|
| Douleurs abdominales |
|
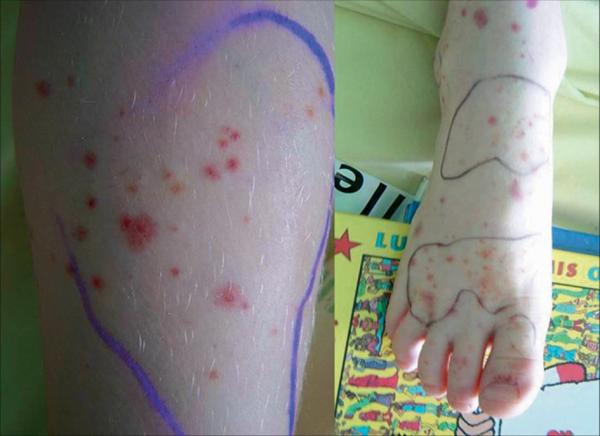
Fig. 24.3. ![]() Purpura rhumatoïde.
Purpura rhumatoïde.
Une atteinte rénale associée doit systématiquement être recherchée au diagnostic, avec réalisation d’une bandelette urinaire à la recherche de protéinurie et/ou d’hématurie, et la mesure de la pression artérielle (existence d’un syndrome néphritique ou néphrotique).
L’atteinte rénale survient dans environ un tiers des cas, le plus souvent dans les 6 premières semaines et avant le 6e mois, rarement avant les atteintes extra-rénales, parfois lors d’une énième poussée. La sur-veillance de la BU est donc indispensable durant les 6 premiers mois ; la constatation d’une protéinurie doit faire réaliser rapidement un rapport protéines sur créatinine urinaires au laboratoire et demander un avis en néphrologie pédiatrique. Elle est la cause de 10 à 15 % des glomérulonéphrites et de 1,5 % des insuffisances rénales terminales de l’enfant.
D’autres complications évolutives, parfois inaugurales, sont à redouter :
- digestives (avant tout) :
- invagination intestinale aiguë (la plus fréquente, voir chapitre 17) (fig. 24.4A) ;
- hématome des parois ;
- péritonite aiguë par vascularite nécrosante ;
- dénutrition ;
- urogénitales : orchite (fig. 24.4B), sténose urétérale (souvent bilatérale) sur urétérite ;
- neurologiques (rares) : céphalées, convulsions, hémorragie intracrânienne, vascularite cérébrale ;
- hémorragies pulmonaires (exceptionnelles).
Enquête paraclinique
Examens complémentaires systématiques :
- NFS-plaquettes : plaquettes normales ;
- BU : dépister une atteinte rénale (protéinurie, hématurie) ;
- créatininémie avec calcul du débit de filtration glomérulaire (formule de Schwartz chez l’enfant).
Autres examens selon les cas :
- échographie abdominale : indiquée en cas de fortes douleurs abdominales ; elle peut mettre en évi-dence des hématomes pariétaux, un boudin d’invagination ;
- bilan hépatique et lipasémie en cas d’atteinte digestive ;
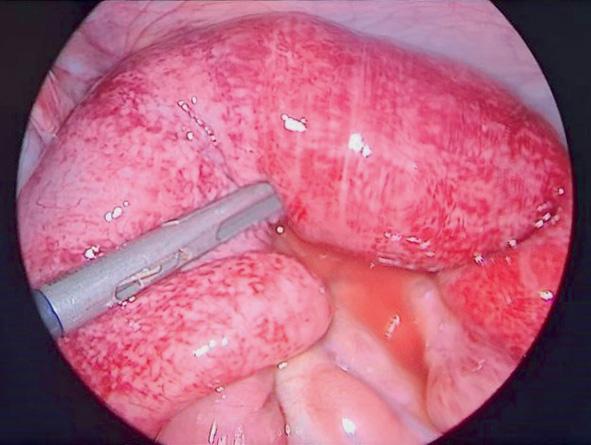
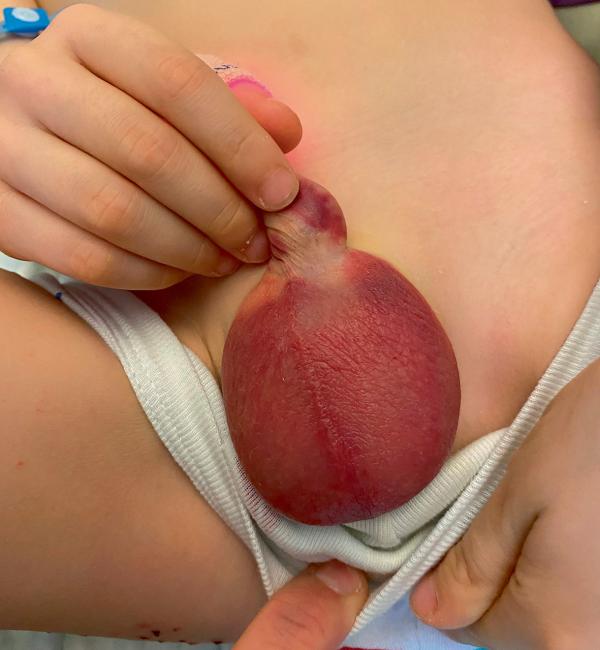
Fig. 24.4. ![]() Complications d’un purpura rhumatoïde.
Complications d’un purpura rhumatoïde.
A. Invagination intestinale aiguë avec purpura visible sur la muqueuse digestive. B. Orchite.
Source : 24.4 A Sabine Irtan et 24.4 B Grégoire Benoist.
- protéinurie, créatininurie, ± dosage de l’albumine sur échantillon urinaire en cas de suspicion d’atteinte rénale (BU positive en protéinurie et/ou hématurie), et avis spécialisé pour discuter la réa-lisation d’une ponction-biopsie rénale ;
- biopsie cutanée : pratiquée exceptionnellement en cas d’incertitude diagnostique (purpura prolongé sur plusieurs mois, présentation atypique) pour éliminer un diagnostic différentiel (périartérite noueuse, vascularite à ANCA, etc.).
3. Prise en charge
Traitement en urgence
La prise en charge est habituellement ambulatoire.
Le traitement est symptomatique : antalgiques (AINS contre-indiqués en cas de glomérulonéphrite ou de saignement gastro-intestinal actif), antispasmodiques. Le repos est sans effet sur la prévention des complications et sur le pronostic.
L’hospitalisation est parfois nécessaire en cas de complications (surtout abdominale ou rénale avec nécessité d’avis spécialisé). Le recours aux corticoïdes peut alors se discuter dans certaines situations qui sont toutefois peu fréquentes (atteinte sévère d’organe, orchite…).
Évolution
Le pupura rhumatoïde est le plus souvent bénin, la poussée étant généralement résolutive spontané-ment en 2 à 6 semaines.
Le suivi est essentiellement clinique avec réalisation d’une bandelette urinaire (recherche de protéinurie) et mesure de la pression artérielle régulièrement pendant au moins 6 mois, même en cas de norma-lité au diagnostic.
En cas d’atteinte atypique par son intensité, ses signes accompagnateurs ou sa durée prolongée, un avis spécialisé est recommandé (dermatologie ou rhumatologie pédiatrique).
Purpura rhumatoïde = cause de purpura vasculaire fréquente chez l’enfant.
Triade = purpura vasculaire, manifestations articulaires, douleurs abdominales.
Pronostic = atteinte rénale, surveillance de la BU.
B. Purpura thrombopénique immunologique (PTI) aigu
1. Généralités
C’est une cause de purpura thrombopénique assez fréquente chez l’enfant.
Le pic d’âge de survenue est entre 2 et 5 ans.
Le PTI est lié à la destruction périphérique des plaquettes, par un processus immun.
Des facteurs déclenchants sont parfois retrouvés : rhinopharyngite, vaccination récente (ROR), médicament.
C’est un diagnostic d’élimination, qui pour être porté, nécessite d’éliminer les autres causes de thrombopénie.
2. Diagnostic
Enquête clinique
Le PTI se présente comme un syndrome hémorragique isolé, avec purpura cutanéomuqueux (fig. 24.5).
À l’anamnèse :
- absence d’antécédents évocateurs de thrombopénie constitutionnelle ;
- absence de signes associés évocateurs d’hémopathie maligne ou de SHU ou de sepsis ;
- absence de signes associés évocateurs de maladie auto-immune de type lupus : arthralgies, arthrite, asthénie, autre éruption cutanée ;
- absence de signes orientant vers un déficit immunitaire héréditaire : infections à répétition et/ou in-habituellement sévère(s).
À l’examen physique :
- appréciation de la sévérité du syndrome hémorragique : score de Buchanan ;
- recherche d’autres signes cliniques de gravité (mentionnés précédemment) ;
- absence de syndrome tumoral (hémopathie), d’HTA (SHU) ou de sepsis.
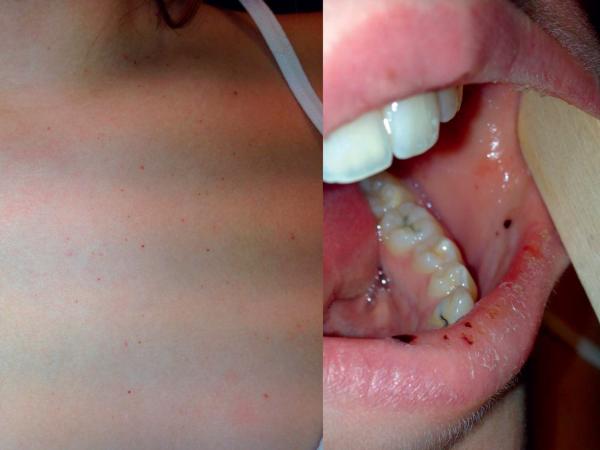
Fig. 24.5. ![]() Purpura thrombopénique immunologique : pétéchies cutanées et buccales.
Purpura thrombopénique immunologique : pétéchies cutanées et buccales.
Enquête paraclinique
Examens complémentaires absolument nécessaires :
- NFS-plaquettes, réticulocytes : thrombopénie isolée, pas d’atteinte des autres lignées (sinon évoquer une hémopathie maligne), pas de lymphopénie notable (sinon évoquer un déficit immunitaire ou une maladie auto-immune associée, par exemple lupus systémique) ;
- frottis sanguin : pas de cellules anormales (blastes : en faveur d’une leucémie aiguë), morphologie plaquettaire normale, absence de schizocytes ;
- hémostase avec TP, TCA, fibrinogène (écarter une CIVD) ;
- BU (protéinurie et hématurie), créatininémie (écarter un SHU) ;
- groupe (deux déterminations), Rhésus, RAI (en vue d’une éventuelle transfusion).
Autres examens complémentaires recommandés en fonction du contexte :
- test de Coombs érythrocytaire, haptoglobine et bilirubine : en cas d’anémie et/ou taux de réticulo-cytes élevé en faveur d’une hémolyse ;
- scanner cérébral et fond d’œil : en cas de céphalées et/ou signes neurologiques anormaux ;
- imagerie abdominale : en cas de syndrome abdominal aigu (vomissements, subocclusion, méléna, ménométrorragies, traumatisme) ;
- dosage pondéral des immunoglobulines et sérologies virales en fonction du contexte (VIH, VHB, VHC), et prélèvement de sérothèques (en prévision du traitement par Ig) ;
- facteurs antinucléaires (FAN) (surtout si âge > 8 ans) ± anti-ADN natif et antigènes solubles ; dosage du complément (C3, C4, CH50) à la recherche de lupus (surtout si lymphopénie ou anémie hémoly-tique auto-immune associée) ;
- phénotypage lymphocytaire : en cas d’argument pour un DIH.
Myélogramme :
- indiqué en cas de doute diagnostique, en particulier si bicytopénie ou signes en faveur d’une hémo-pathie maligne (douleurs osseuses, syndrome tumoral clinique, cellules anormales au frottis sanguin) ;
- indiqué pour certains experts (afin d’exclure une hémopathie maligne) si décision de débuter une corticothérapie sans évaluation préalable par un clinicien senior et un cytologiste expérimentés pou-vant confirmer la normalité de l’examen clinique et de la NFS avec frottis sanguin (différable de 12 heures si traitement débuté en garde) ;
- examen non contre-indiqué par la thrombopénie, réalisé sous antalgiques et prémédication ;
- aspect : moelle riche, avec présence de mégacaryocytes en quantité normale voire augmentée, sans cellules malignes.
Bilan ou actes contre-indiqués ou à différer :
- PL ;
- tout geste potentiellement invasif en dehors du myélogramme (si indiqué) : injection IM, prise de température rectale…
3. Prise en charge
Modalités thérapeutiques
L’abstention thérapeutique se justifie en l’absence de signes de gravité clinique.
En cas de saignement modéré, il peut être proposé une corticothérapie per os ou des immunoglobulines polyvalentes IV. En cas d’hémorragies sévères et/ou prolongées, le traitement associe des immunoglobulines polyvalentes IV et une corticothérapie IV.
Selon la sévérité du syndrome hémorragique et la tolérance de l’anémie, une transfusion de culots globulaires rouges peut être nécessaire. L’arrêt de tout saignement actif est la priorité.
Une transfusion de plaquettes est d’indication exceptionnelle, les plaquettes étant en général très rapidement détruites. Elle peut être indiquée en cas de pronostic vital engagé (choc hémorragique, hémorragie intracrânienne, par exemple) pour passer un cap.
Évolution
Le PTI aigu de l’enfant est habituellement de bon pronostic.
La recherche de signes hémorragiques, en particulier au niveau de la cavité buccale, doit être faite lors de toute consultation médicale et de manière quotidienne par la famille au décours.
Le contrôle de la numération plaquettaire est fonction de la clinique, et s’effectue au moins 48 heures après le début du traitement de première intention quand il a eu lieu.
La guérison définitive survient dans la majorité des cas en quelques jours ou semaines.
Il existe un risque de récidive ainsi que de rares cas d’évolution chronique nécessitant un suivi spécialisé en hématologie pédiatrique.
Le PTI peut révéler un déficit immunitaire primitif ou un lupus systémique juvénile, qui doit être recherché surtout chez des patients qui présentent un PTI en dehors de l’âge « habituel » et/ou avec une évolution chronique
PTI aigu = purpura thrombopénique non rare chez l’enfant.
Myélogramme : indiqué surtout en cas de doute diagnostique.
Transfusion de plaquettes d’indication exceptionnelle.
Références

|
HAS. Purpura thrombopénique immunologique de l’enfant et de l’adulte. Protocole national de diagnostic et de soins. 2017. https://www.has-sante.fr/upload/docs/application/pdf/2017-06/dir36/pnds… |

|
SHIP. Purpura thrombopénique idiopathique de l’enfant : mesures d’accompagnement. 2007. http://www.cerevance.org/website/datadev/article/file/17546325812912420… |