Items, objectifs pédagogiques
Item 297 – Cancer de l’enfant : particularités épidémiologiques, diagnostiques et thérapeutiques
- Expliquer les particularités épidémiologiques et diagnostiques des principaux cancers de l’enfant.
Item 315 – Leucémies aiguës
- Éléments cliniques et de diagnostic d’une leucémie aiguë (hors classification).
Avant de commencer…
Cet item exige de connaître les particularités épidémiologiques et diagnostiques des cancers de l’enfant. Il n’a pas pour objectif d’évaluer leur prise en charge thérapeutique.
Quatre exemples de cancers fréquents chez l’enfant seront développés : leucémie aiguë, tumeurs cérébrales, tumeurs abdominales (neuroblastome, néphroblastome) et tumeurs osseuses.
Les circonstances et signes cliniques devant faire évoquer une tumeur thoracique chez l’enfant sont évoqués dans le chapitre 59 « Opacités et masses intrathoraciques ».
Les cancers de l’enfant sont des maladies rares, essentiellement sporadiques même si 10 % sont actuellement rapportés à une prédisposition génétique.
Dans les pays industrialisés, ils représentent la deuxième cause de mortalité entre 1 et 15 ans.
Le taux de guérison global des cancers de l’enfant est d’environ 80 %. Les objectifs actuels de la cancérologie pédiatrique sont d’augmenter encore les taux de guérison, tout en diminuant les risques de séquelles.
Les types histologiques sont très différents de ceux des cancers de l’adulte.
Les hémopathies malignes (leucémies et lymphomes) sont la première cause de cancer de l’enfant.
Les autres cancers fréquents sont les tumeurs cérébrales, les tumeurs embryonnaires (néphroblastome, neuroblastome), les sarcomes osseux (ostéosarcome, sarcome d’Ewing) ou des tissus mous (rhabdo-myosarcomes).
Les signes d’appel des cancers de l’enfant sont nombreux et souvent très peu spécifiques, sans altération de l’état général, alors que ce sont des urgences compte tenu de leur évolutivité très rapide.
Les circonstances diagnostiques peuvent être en rapport avec des signes directement liés à la tumeur et/ou à des signes indirects de compression ou d’envahissement.
Le diagnostic repose sur la convergence d’arguments cliniques et d’imagerie, et le plus souvent sur des éléments anatomopathologiques et de caractérisation biologique de la tumeur.
I. Particularités épidémiologiques
A. Prévalence
Les cancers de l’enfant représentent moins de 1 % de l’ensemble des cancers.
Ce sont des maladies rares : environ 2 500 cas par an en France entre les âges de 0 et 18 ans, dont 1 700 cas par an survenant avant l’âge de 15 ans parmi lesquels 50 % des cas avant l’âge de 5 ans. Il existe une prédominance masculine (sexratio = 1,2).
Les taux de guérison des cancers de l’enfant atteignent aujourd’hui 80 % dans les pays industrialisés grâce à leur prise en charge pluridisciplinaire par des équipes hautement spécialisées et à leur grande sensibilité aux médicaments de chimiothérapie cytotoxiques.
De grands progrès thérapeutiques ont été réalisés au cours des dernières années, grâce aux acquis de stratégies thérapeutiques développées dans le cadre de protocoles de recherche clinique multicentriques et internationaux.
Leur pronostic est donc globalement meilleur que celui de la plupart des cancers survenant chez les adultes.
B. Prédispositions génétiques et autres facteurs de risque
Les cancers de l’enfant sont essentiellement sporadiques.
Peu de facteurs environnementaux connus sont impliqués dans leur genèse. On peut citer les irradiations à fortes doses, des virus tels que EBV (lymphomes de Burkitt, carcinome indifférencié du nasopharynx, maladie de Hodgkin), VIH (lymphomes, léiomyosarcome) ou VHB (hépatocarcinome) et certains facteurs iatrogènes bien établis comme les traitements alkylants et les inhibiteurs des topoisomérases (leucémie) ou les traitements immunosuppresseurs (lymphome).
Une proportion des cancers de l’enfant, actuellement de 10 %, survient dans le cadre de syndromes connus de prédisposition génétique, tels que les formes héréditaires du rétinoblastome (RB1) (risque accru de second cancer), le syndrome de Li-Fraumeni (TP53) (tumeurs des tissus mous et des os, lymphomes, tumeurs du SNC, corticosurrénalomes), le syndrome de Beckwith-Wiedemann (WT2) (néphroblastomes, hépatoblastomes), la trisomie 21 (leucémies), les neurofibromatoses de types 1 et 2 (NF1 et 2) (tumeurs du SNC), et d’autres syndromes plus rares encore.
La reconnaissance d’un syndrome de prédisposition est importante pour l’enfant et sa famille. En effet, certains de ces syndromes entraînent un risque de toxicité importante des traitements, justifiant des aménagements dans les modalités thérapeutiques (non-recours à la radiothérapie ou à certaines chimiothérapies, par exemple), les doses ou les modalités de surveillance. À l’échelon familial, l’enquête génétique peut permettre de mettre en place des mesures de surveillance particulière pour les enfants porteurs de l’anomalie génétique, voire de proposer un diagnostic pré-natal pour les grossesses à venir.
Ainsi, il faut savoir évoquer un syndrome de prédisposition devant certaines situations de découverte d’un cancer chez l’enfant. Certaines sont liées au cancer lui-même (histoire familiale de cancers, types de cancers rares, survenue de cancers synchrones ou métachrones) ; d’autres sont liées à l’association à d’autres anomalies, en particulier développementales (dysmorphies faciales ou autres anomalies congénitales, défaillance intellectuelle, troubles de la croissance, anomalies de la peau ou de l’hématopoïèse).
Cancers de l’enfant : 2 500 cas/an en France de 0 à 18 ans, taux de guérison d’environ 80 %.
C. Répartition statistique
Les types histologiques et la fréquence des cancers de l’enfant sont très distincts de ceux de l’adulte.
Schématiquement : 60 % des cancers de l’enfant sont des tumeurs solides non hématologiques, 40 % des hémopathies malignes (leucémies, lymphomes). Leur répartition diffère selon l’âge (fig. 27.1 ; les chiffres exacts ne sont pas à mémoriser).
Avant l’âge de 5 ans, les cancers prédominants sont les leucémies aiguës, les tumeurs embryonnaires (dites du blastème) spécifiques de l’enfant (rétinoblastomes, neuroblastomes, néphroblastomes, hépatoblastomes), certaines tumeurs cérébrales (médulloblastomes, gliomes de bas grade) et quelques sarcomes (rhabdomyosarcomes).
Après l’âge de 10 ans, il s’agit surtout de tumeurs cérébrales (gliomes, médulloblastomes), de sarcomes osseux (ostéosarcomes, sarcome d’Ewing) et des tissus mous (rhabdomyosarcomes et non-rhabdomyosarcomes), de lymphomes (hodgkiniens et non hodgkiniens), et des tumeurs germinales malignes (principalement gonadiques à cet âge).
Cancers de l’enfant les plus fréquents : leucémies aiguës, tumeurs cérébrales, lymphomes, neuroblastomes, néphroblastomes.
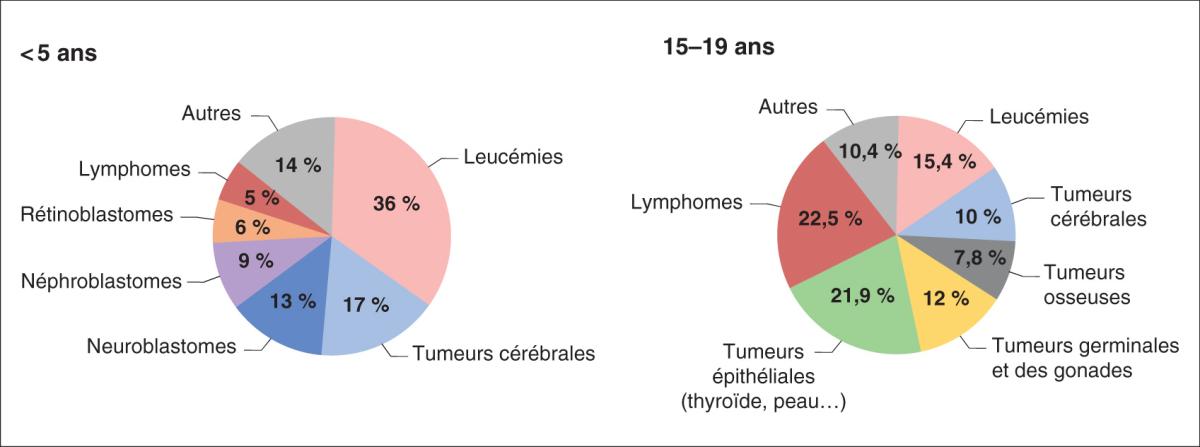
Fig. 27.1. ![]() Proportion des différents types de cancers de l’enfant et de l’adolescent selon l’âge.
Proportion des différents types de cancers de l’enfant et de l’adolescent selon l’âge.
II. Particularités diagnostiques
A. Être vigilant
Les signes d’appel de cancers de l’enfant doivent être reconnus le plus tôt possible.
En effet, la précocité du diagnostic permet souvent le recours à des traitements moins agressifs. Toutefois, la rareté et la rapidité évolutive de ces cancers ne permettent pas d’envisager un dé-pistage systématique, sauf dans de rares cas de syndromes de prédisposition.
Un interrogatoire attentif prenant en compte les symptômes décrits, un examen clinique exhaustif ainsi qu’une analyse vigilante de l’évolution permettent une stratégie diagnostique raisonnable pour l’identification ou la réfutation de signes évocateurs de cancers, tout en prenant en compte l’inquiétude familiale.
Les signes d’appel des cancers de l’enfant sont nombreux.
Ils peuvent être découverts de manière fortuite par les parents (masse abdominale au cours de la toilette) ou par le médecin traitant (examen systématique) ou du fait de la persistance sous traitement bien conduit, d’un signe ou d’un symptôme d’allure initialement bénigne.
Certains signes ou symptômes généraux peuvent survenir mais sont beaucoup plus rares qu’au cours des cancers de l’adulte : fatigue isolée, diminution de l’appétit, amaigrissement ou mauvaise prise staturo-pondérale chez le jeune enfant, infections anormalement répétées.
Les signes d’appel sont souvent banals et malheureusement parfois « banalisés ».
Ils sont le plus souvent d’évolution rapide, survenant chez un enfant dont l’état général demeure conservé, et peuvent parfois mettre en jeu du fait de la localisation tumorale, le pronostic fonctionnel ou vital immédiat.
Tout symptôme ou signe persistant et fixe pendant plusieurs jours, toute douleur nocturne (céphalées, douleurs abdominales ou des membres), insolites ou durables, doivent faire évoquer une étiologie organique qui peut être parfois cancéreuse.
Signes d’appel multiples et parfois banals… à ne pas banaliser.
B. Circonstances diagnostiques
Les signes peuvent être en rapport avec la découverte « directe » de la tumeur :
- masse abdominale :
- intrapéritonéale : lymphome de Burkitt, hépatoblastome ;
- rétropéritonéale : néphroblastome, neuroblastome ;
- abdominopelvienne : tumeur germinale maligne, neuroblastome, sarcome ;
- tuméfaction des membres ou des parois révélant des sarcomes :
- des tissus mous : le plus souvent rhabdomyosarcome ;
- osseux : ostéosarcome, tumeur d’Ewing ;
- adénopathies « froides » (dures et adhérentes, souvent sans inflammation ni douleur) :
- leucémies ;
- lymphomes ;
- autres signes :
- reflet blanc pupillaire (leucocorie) : rétinoblastome ;
- augmentation du volume scrotal : rhabdomyosarcome, tumeur germinale maligne, lym-phome ;
- hématurie : néphroblastome ;
- masse péri-orificielle ou intra-orificielle ± saignement : rhabdomyosarcome, tumeur germinale maligne vaginale.
Les symptômes peuvent être en rapport avec des signes « indirects » de la tumeur :
- HTIC, signes neurologiques déficitaires : tumeur cérébrale ;
- compression médullaire (y penser chez un enfant douloureux et difficilement mobilisable) : tumeurs osseuses rachidiennes, neuroblastome, hémopathies malignes, tumeurs médullaires ;
- dyspnée par compression médiastinale : lymphome (le plus souvent non hodgkinien), rare-ment tumeur germinale maligne, sarcomes ou neuroblastome ;
- obstruction respiratoire haute, troubles de déglutition : tumeur ORL (lymphome, rhabdo-myosarcome) ;
- protrusion oculaire : tumeur orbitaire primitive (rhabdomyosarcome) ou métastase orbitaire (neuroblastome, hémopathie maligne) ;
- douleurs osseuses localisées : tumeurs osseuses (ostéosarcome, tumeur d’Ewing) ;
- douleurs osseuses diffuses ± boiterie : atteintes de la moelle osseuse (leucémie, neuroblas-tome ou sarcomes métastatiques) ;
- difficultés d’émission d’urines ou de selles : tumeurs abdominopelviennes (sarcome, neuro-blastome, tumeurs germinales malignes), tumeurs intracanalaires ;
- strabisme : rétinoblastome ;
- prurit : lymphome de Hodgkin.
Plus rarement, le cancer peut se révéler au cours de certaines situations d’urgence :
- dyspnée asphyxiante : lymphome non hodgkinien médiastinal, tumeur ORL ;
- pancytopénie (anémie aiguë, saignement, fièvre), CIVD : hémopathie ;
- HTIC d’évolution rapide avec signes neurovégétatifs, convulsions : tumeur cérébrale ;
- fracture osseuse pathologique : tumeur osseuse ;
- hémorragie intra-abdominale : néphroblastome ;
- hypercalcémie : origine paranéoplasique ou lyse osseuse.
Alerte : HTIC, boiterie et douleurs osseuses, masse abdominale, strabisme, leucocorie.
C. Démarche diagnostique
1. Conduite générale
Le diagnostic est habituellement porté sur une convergence d’arguments cliniques et paracliniques.
La démarche diagnostique vise à confirmer le diagnostic, préciser la nature et les caractéristiques de la tumeur, ainsi qu’à établir le bilan d’extension locorégional et général.
Les éléments demandés sont fonction des hypothèses étiologiques : examens biologiques (NFS-plaquettes, examens biochimiques sanguins standards, marqueurs tumoraux), examens d’imagerie, examens anatomopathologiques et de caractérisation biologique de la tumeur.
La stratégie diagnostique doit être rigoureuse et organisée en collaboration avec un centre de référence en cancérologie pédiatrique, afin d’éviter tout préjudice concernant la prise en charge thérapeutique et le pronostic
2. Spécificités à connaître
Principaux marqueurs tumoraux particuliers au diagnostic des tumeurs pédiatriques :
- α-fœtoprotéine (AFP) : tumeurs germinales malignes (composante « tumeur du sac vitellin »), hépatoblastome ;
- β-hCG : tumeurs germinales malignes (composante « choriocarcinome ») ;
- métabolites des catécholamines et dopamine urinaires : neuroblastome.
Autres marqueurs moins spécifiques souvent recherchés :
- hypercalcémie : atteintes ostéomédullaires étendues, tumeurs rhabdoïdes ;
- élévation des LDH (marqueur d’agressivité) : lymphome malin non hodgkinien (LMNH), neuroblastome métastatique.
Les examens d’imagerie permettent le diagnostic topographique des lésions, contribuent souvent au diagnostic étiologique et participent au bilan d’extension.
Techniques d’imagerie utilisées suivant les situations :
- examens radiographiques standards (très souvent contributifs) :
- radiographie de thorax ;
- radiographies de segments osseux : en cas de douleurs ou de tuméfactions localisées ;
- ASP (rarement) : en cas de tumeur abdominale ;
- échographie ± doppler (très précieuse) :
- précise le caractère intra- ou rétropéritonéal d’une tumeur abdominale ;
- précise l’origine d’une tumeur abdominopelvienne ;
- contribue à l’exploration des lésions cervicales, des membres et des parois ;
- examens plus complexes (en fonction des résultats des premières explorations) :
- TDM ;
- IRM : tumeur du SNC, tumeurs osseuses ou des tissus mous ou toute autre tumeur en fonction de son accessibilité par rapport à la TDM ;
- scintigraphie au technétium : tumeurs osseuses primitives et/ou métastatiques ;
- scintigraphie au méta-iodobenzylguanidine (MIBG) : neuroblastome ;
- tomographie par émission de positons (TEP) ± TDM : validée dans l’exploration des lymphomes hodgkiniens et non hodgkiniens mais également de plus en plus utilisée dans d’autres types tumoraux (en particulier les sarcomes).
Examens anatomopathologiques avec des explorations :
- cytologiques :
- myélogramme : leucémie ;
- ponction ganglionnaire ou tumorale : certaines formes de LMNH ;
- histologiques : abords biopsiques percutanés ou chirurgicaux a minima.
Les examens de caractérisation biologique de la tumeur sont de plus en plus souvent indispensables pour préciser le diagnostic et mieux définir le pronostic.
Ils nécessitent généralement une congélation tumorale, qu’il faut prévoir lors des prélèvements.
Contact rapide avec un centre de référence en cancérologie pédiatrique.
Bilan paraclinique à visée diagnostique et pronostique.
D. Annonce diagnostique
1. Principes généraux
L’annonce d’une maladie grave est un moment particulier de la relation médecin-malade, en l’occurrence entre le médecin, l’enfant et ses parents. Elle conduit à un retentissement psychologique majeur chez l’enfant et sa famille.
C’est ainsi un temps essentiel pour créer une alliance thérapeutique entre l’enfant, ses parents, et les équipes médicales impliquées dans la démarche diagnostique et la prise en charge thérapeutique (service de pédiatrie, centre de référence en cancérologie pédiatrique, service de chirur-gie infantile spécialisée), alliance indispensable à la réalisation de soins de qualité.
Délivrée parfois par le médecin traitant qui a évoqué le diagnostic, cette annonce doit être re-layée par les centres de référence en cancérologie pédiatrique selon un « dispositif » prévu dans le cadre du plan cancer.
L’annonce du diagnostic de cancer demande du temps, de la disponibilité et de l’expérience.
Elle est délivrée de manière progressive au cours d’entretiens répétés et rapprochés, réalisés avec empathie et dans un lieu permettant confidentialité et intimité.
Elle est faite dans le respect de la personnalité de l’enfant et sa famille, de leurs attentes et be-soins, en tenant compte du contexte culturel et psychologique. Elle doit être adaptée à l’âge et au niveau de compréhension de l’enfant.
2. Dispositif d’annonce
Le dispositif d’annonce hospitalier est ainsi construit autour de quatre temps, souvent menés en partie en parallèle. Chacun de ces temps peut nécessiter plusieurs entretiens successifs.
- Temps médical :
- annonce du diagnostic puis des propositions et décisions débattues en réunion de concertation pluridisciplinaire (RCP) ;
- présentation des risques à court, moyen et long terme des options thérapeutiques et justification de l’option choisie avec proposition de recours à un deuxième avis médical ;
- proposition d’inclusion le cas échéant dans un essai thérapeutique ou une étude interventionnelle ou non interventionnelle, selon les critères d’éligibilité ;
- finalisation du projet sous forme d’un programme personnalisé de soins (PPS), nécessitant le plus souvent la mise en place d’un cathéter central de longue durée.
Il est nécessaire de valider au fur et à mesure la compréhension de l’information délivrée. La re-mise de documents écrits sur la maladie et ses traitements est un support souvent utile à une in-formation de qualité mais ne dispense en aucune façon des entretiens successifs.
L’aide des associations de parents est souvent également utile, soit par des contacts directs, soit par le recours à des documents d’information sur ces associations.
- Temps d’accompagnement soignant :
- précision des modalités thérapeutiques, visite du service ;
- identification en équipe pluriprofessionnelle des besoins d’accompagnement social et psychologique.
- Temps d’accès aux soins de support (psychologue, assistante sociale, kinésithérapeute…). L’enfant bénéficie d’une prise en charge à 100 %. Un projet d’accueil individualisé (PAI) permet une scolarité adaptée à l’état de santé de l’enfant.
- Temps d’articulation avec la médecine de ville (information du médecin traitant) et l’équipe de pédiatrie de proximité.
Annonce diagnostique avec information claire et complète. Alliance thérapeutique.
III. Points clés sur certains cancers de l’enfant
A. Leucémie aiguë
1. Généralités
Les leucémies aiguës sont les cancers les plus fréquents de l’enfant (30 %).
Ce sont des proliférations malignes de précurseurs de cellules sanguines bloqués à un stade précoce de leur différenciation (blastes).
On en distingue deux variétés suivant le type cytologique des cellules blastiques :
- les leucémies aiguës lymphoblastiques (LAL) ;
- leucémies aiguës myéloïdes (LAM).
Les LAL représentent environ 80 % des leucémies aiguës de l’enfant.
On dénombre 350 à 400 nouveaux cas par an en France, avec deux pics de fréquence : 2 à 5 ans (principal pic) et adolescence.
Schématiquement : 75 % sont des proliférations lymphoïdes de cellules immatures de la lignée B, 20 % sont des proliférations de la lignée T. Il existe de très rares proliférations de cellules lymphoblastiques B matures ; il s’agit de leucémies de Burkitt.
Les leucémies aiguës doivent être prises en charge dans un centre spécialisé qui affirmera le diagnostic, assurera le dépistage et le traitement des complications initiales et réalisera les explorations spécialisées permettant de caractériser la leucémie et définir le PPS en fonction des facteurs pronostiques mis en évidence.
2. Diagnostic
Signes cliniques d’appel
La symptomatologie est le plus souvent aspécifique, évoluant typiquement depuis 2 à 3 semaines et témoignant de l’insuffisance médullaire et/ou de l’infiltration blastique.
Manifestations d’insuffisance médullaire :
- anémie : asthénie, pâleur, éventuelle dyspnée ;
- thrombopénie : syndrome hémorragique cutanéomuqueux, purpura pétéchial, ecchymoses diffuses ou dans des sites inhabituels, épistaxis, gingivorragies, hématurie, sang dans les selles ;
- neutropénie : fièvre prolongée, angine récidivante ou ne cédant pas aux antibiotiques, aphtose.
Manifestations du syndrome tumoral liées à la prolifération leucémique dans la moelle osseuse et les organes hématopoïétiques secondaires et lymphoïdes :
- douleurs osseuses, boiterie (enfant ne voulant plus monter les escaliers, voulant être porté, enfant « trop sage », immobile sur son lit, qui ne joue plus…) ;
- hépatomégalie, splénomégalie, adénopathies périphériques, amygdalomégalie, gros testicule, infiltration des gencives ;
- gêne respiratoire voire syndrome cave supérieur en cas de syndrome tumoral médiastinal ;
- signes neurologiques périphériques (compression médullaire sur tassement vertébral voire envahissement tumoral).
Signes de complications aiguës à repérer et prendre en charge en urgence :
- détresse respiratoire : compression médiastinale, épanchement pleuropéricardique, syn-drome de leucostase pulmonaire (obstruction capillaire, HTAP, œdème pulmonaire) ;
- CIVD : en cas de leucémie hyperleucocytaire (> 100 Giga/l) ou LAM promyélocytaire ;
- insuffisance rénale : par infiltration rénale ou syndrome de lyse tumorale (leucémie hyperleu-cocytaire ou leucémie de Burkitt) ;
- troubles de la vigilance, céphalées : leucostase cérébrale (leucémies hyperleucocytaires).
Stratégie diagnostique
L’hémogramme (NFS-plaquettes) peut donner les résultats suivants :
- forme hyperleucocytaire avec blastose sanguine d’importance variable ;
- forme pancytopénique sans blastes ;
- parfois normal, ce qui n’élimine pas le diagnostic.
Le diagnostic ne peut et ne doit être porté qu’après réalisation d’un myélogramme :
- cytologie médullaire au microscope : > 20 % de cellules blastiques ;
- examen immunohistochimique : coloration par la myéloperoxydase et les estérases, négatives dans les LAL ;
- classification selon la morphologie pour les LAL (L1, L2, L3) et le degré de différenciation pour les LAM (M0 à M7).
Ce myélogramme est complété par les examens suivants :
- immunophénotypage sur cellules médullaires en suspension en cytométrie de flux :
- confirmation du caractère hématopoïétique de la prolifération (CD34) ;
- pour les LAL : caractère B ou T selon les antigènes positifs ;
- pour les LAM : confirmation du caractère myéloïde ;
- génétique hématologique médullaire = anomalies présentes uniquement dans les blastes (ces caractéristiques, associées à la réponse précoce au traitement, sont les principaux éléments pronostiques des leucémies de l’enfant) :
- ploïdie des cellules blastiques ;
- ou anomalies structurales : délétions, inversions péricentriques, duplications, translo-cations ;
- ± compléments de lecture par techniques de fluorescence par hybridation in situ (FISH) ;
- examen en biologie moléculaire à la recherche de transcrits de fusion récurrents (par exemple, BCR-ABL) ou autres anomalies génomiques (de plus en plus par techniques de séquençage à haut débit).
Enfin, d’autres examens sont nécessaires au diagnostic et à la prise en charge :
- examen du LCS : recherche d’un envahissement neuroméningé initial (présence de blastes lors du décompte cellulaire et sensibilisation par technique de concentration Cytospin®) ; at-tention : l’atteinte méningée est asymptomatique dans l’extrême majorité des cas et ne peut être mise en évidence que par une analyse du LCS par ponction lombaire ; on peut néanmoins retrouver des atteintes des paires crâniennes (paralysie du VII le plus souvent) ;
- examens d’imagerie non systématiques à prévoir en fonction du tableau initial :
- radiographie de thorax, échographie abdominale et/ou testiculaire ;
- IRM cérébrale ;
- bilan préthérapeutique :
- bilan prétransfusionnel ;
- syndrome de lyse : hyperuricémie, hyperkaliémie, hypocalcémie, hyperphosphorémie.
L’ensemble de ces caractéristiques permettent de proposer une polychimiothérapie personnalisée conduisant à la guérison de plus de 80 % des enfants et adolescents âgés de 1 an et 18 ans et atteints de LAL. Les protocoles de LAL actuels tiennent ainsi compte de facteurs cliniques initiaux (âge < 1 an et > 10 ans, atteinte neuroméningée), biolo-giques (blastose sanguine > 50 000/mm3, immunophénotype), génétiques (anomalies du clone blastique) et de réponse précoce au traitement (corticosensibilité à J8, rémission cytologique en fin d’induction, décroissance rapide de la maladie résiduelle mesurée par PCR ou cytométrie de flux) pour la stratification du traitement.
B. Tumeurs cérébrales
1. Généralités
Les tumeurs cérébrales de l’enfant représentent la première cause de tumeurs solides, avec environ 400 nouveaux cas par an en France avant l’âge de 15 ans.
La moitié sont des tumeurs supratentorielles (astrocytomes de bas grade, craniopharyngiomes, gliomes des voies optiques…) et l’autre moitié des tumeurs de la fosse postérieure (médulloblastomes, tumeurs malignes du tronc cérébral…).
Beaucoup de tumeurs cérébrales restent de pronostic sombre en raison de leur faible chimiosensibilité et de l’importance et de la difficulté du traitement local chirurgical ou radiothérapique.
De plus, les effets secondaires des traitements sur un système nerveux en développement sont importants. Ils nécessitent un suivi multidisciplinaire (endocrinologique, sensoriel, orthopédique, neurologique, psychologique, etc.), et contre-indiquent l’utilisation de la radiothérapie chez les moins de 5 ans.
2. Diagnostic
Signes cliniques d’appel
L’hypertension intracrânienne (HTIC) est le signe d’appel le plus fréquent, se manifestant typiquement par des céphalées intenses (classiquement matinales) et des vomissements.
Les formes frustes sont fréquentes (céphalées fluctuantes et peu intenses, sans horaire particulier, sans vomissement associé) et les formes trompeuses également (nausées/vomissements isolés, douleurs abdominales, irritabilité, fléchissement scolaire), les céphalées étant absentes ou passant au deuxième plan, avec un tableau pouvant égarer vers des troubles d’origine psychologique.
Tout symptôme anormal ou persistant doit mener à un examen neurologique attentif et l’imagerie cérébrale en urgence est toujours demandée en présence d’anomalie clinique.
L’HTIC est très souvent isolée et il existe des faux négatifs fréquents de la recherche d’œdème papillaire au fond d’œil. Toute suspicion d’HTIC doit donc mener à une imagerie cérébrale en urgence, de préférence IRM mais une TDM est souvent réalisée en première intention selon l’accessibilité.
Chez les nourrissons, l’HTIC peut se manifester par une macrocéphalie évolutive, une fontanelle bombée, une régression des acquisitions psychomotrices et, tardivement, par un « regard en coucher de soleil » ; l’imagerie cérébrale est également une urgence devant ces symptômes ; l’échographie transfontanellaire ou la TDM peuvent être une première étape, mais l’IRM reste in-dispensable.
D’autres manifestations peuvent être révélatrices de tumeurs cérébrales, parfois en dehors de tout signe d’HTIC : déficit visuel, comitialité souvent à type de crises partielles, tout signe de localisation neurologique (une atteinte motrice ou des paires crâniennes, syndrome cérébelleux…) ou encore un ou des déficit(s) endocrinien(s) d’origine centrale.
Enfin, le diagnostic de tumeur cérébrale peut être posé dans le cadre d’un syndrome de prédis-position génétique. Le plus fréquent d’entre eux est la neurofibromatose de type 1, qui prédis-pose aux astrocytomes de bas grade en particulier au niveau des voies optiques ; les autres pré-dispositions sont plus rares et plus souvent diagnostiquées dans le cadre de la découverte de la tumeur cérébrale elle-même que dans un contexte déjà identifié.
Stratégie diagnostique
La découverte d’une tumeur cérébrale impose une prise en charge neuro-oncologique spécialisée pluridisciplinaire et, en cas d’HTIC, un transfert immédiat en neurochirurgie pour réalisation d’une dérivation externe ou interne le plus souvent (ventriculocisternostomie plutôt que par valve de dérivation ventriculopéritonéale). Le recours à la corticothérapie voire aux perfusions IV de mannitol est fréquent.
La démarche diagnostique est réalisée en parallèle de la prise en charge en urgence lorsque celle-ci est nécessaire.
L’IRM cérébrale ou cérébrospinale est l’examen de choix. Elle permet de définir :
• la topographie de la lésion, ses rapports avec les structures fonctionnelles adjacentes ;
• la présence de signes de dissémination tumorale visibles en IRM craniospinale ;
• l’existence d’une dilatation ventriculaire.
Certaines tumeurs peuvent être opérées d’emblée, si possible de façon complète, alors que d’autres seront biopsiées.
Les prélèvements tumoraux doivent faire l’objet d’analyses spécialisées à la fois anatomo-pathologiques (histologie standard, immunohistochimie, hybridation in situ) et moléculaires, au mieux réalisées sur des fragments tumoraux congelés.
Dans les tumeurs de la région suprasellaire ou pinéale, l’analyse des marqueurs (AFP, hCG) dans le sang ou le LCS (souvent alors prélevé dans le cadre d’une chirurgie de dérivation) peut faire le diagnostic de tumeur germinale intracrânienne sécrétante.
Le diagnostic est parfois porté sur une convergence d’arguments cliniques et d’imagerie sans documentation histologique (fig. 27.2 et fig. 27.3) ; on peut citer les tumeurs des voies optiques, sur-tout dans un contexte de neurofibromatose.
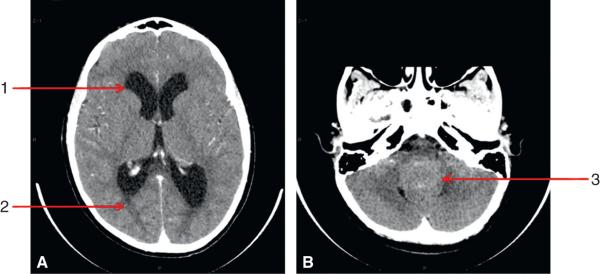
Fig. 27.2. ![]() TDM cérébrale réalisée en urgence devant un tableau d’HTIC.
TDM cérébrale réalisée en urgence devant un tableau d’HTIC.
A. Coupe axiale à l’étage supra-tentoriel : dilatation ventriculaire (1) et résorption trans-épendymaire (2). B. Coupe axiale au niveau de la fosse postérieure : tumeur comblant le 4e ventricule (3).
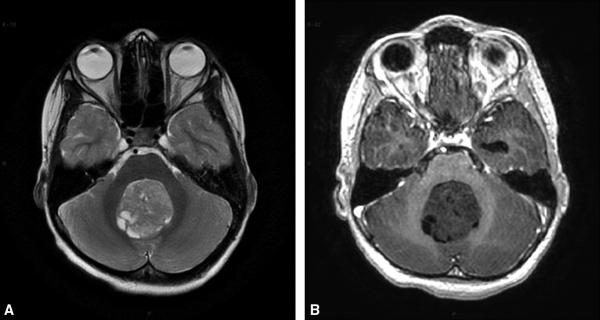
Fig. 27.3. ![]() Méduloblastome.
Méduloblastome.
IRM cérébrale, coupes axiales au niveau de la fosse postérieure. A. T2. B. T1 + gadolinium. Tumeur de la fosse postérieure comblant le 4e ventricule hétérogène prenant peu le contraste. Abord chirurgical initial : diagnostic histologique de médulloblastome.
C. Tumeurs abdominales
1. Généralités
Les circonstances de découverte d’une tumeur abdominale chez l’enfant sont multiples et peuvent être trompeuses.
Parmi les tumeurs les plus fréquentes, on distingue celles de siège :
- rétropéritonéal, rénales ou extrarénales : néphroblastomes, neuroblastomes, tumeurs germi-nales malignes ou tératomes, corticosurrénalomes, phéochromocytome ;
- intrapéritonéal, hépatiques ou extrahépatiques : hépatoblastomes, lymphomes digestifs ;
- pelvien : tumeurs de l’ovaire, tératome sacrococcygien.
On retrouve principalement des tumeurs du blastème avant 5 ans.
Les lymphomes digestifs touchent plutôt l’enfant de 6 à 10 ans avec une prédominance masculine. Les tumeurs ovariennes sont retrouvées à l’adolescence.
2. Diagnostic
Signes cliniques d'appel
Les signes les plus fréquents sont :
- une augmentation du volume abdominal (surtout tumeurs du blastème) ;
- des douleurs abdominales ou une constipation, des troubles urinaires (hématurie évoquant un néphroblastome) ;
- une altération de l’état général (témoignant souvent de métastases ostéomédullaires d’un neuroblastome) ;
- une HTA.
Des signes particuliers orientent d’emblée vers une histologie particulière :
- l’HTA : neuroblastome (par compression vasculo-rénale, par sécrétion d’amines vasopressives, liée à la douleur), phéochromocytome et néphroblastome (d’origine vasculaire) ;
- des signes endocriniens : corticosurrénalome en cas d’hypercorticisme et tumeur des cordons sexuels en cas de pseudopuberté précoce et/ou virilisation ;
- l’existence d’un syndrome opsomyoclonique (mouvements oculaires saccadés, ataxie et mou-vements anormaux) ou d’une diarrhée paranéoplasique (hypersécrétion de VIP), de douleurs osseuses ou fractures sur os pathologique (envahissement ostéomédullaire), d’un hématome péri-orbitaire (syndrome de Hutchinson), de nodules bleutés sous-cutanés enchâssés dans le derme chez le nourrisson : neuroblastome ;
- un syndrome génétique prédisposant (hémihypertrophie corporelle entrant dans un syndrome de Wiedeman-Beckwith) : néphroblastome.
Des tableaux aigus doivent faire évoquer le diagnostic de cancer :
- syndroe abdominal aigu : invagination intestinale aiguë après 5 ans (lymphome de Burkitt), torsion d’annexe (tumeur ovarienne) ;
- rétention aiguë d’urine ou signes moteurs (neuroblastome pararachidien en « sablier ») ;
- choc hypovolémique (rupture tumorale hémorragique).
Stratégie diagnostique
L’imagerie a une place prépondérante dans l’orientation dia-gnostique initiale :
- radiographie standard : l’ASP a un apport diagnostique faible même s’il peut mettre en évidence des calcifications, des plages d’ostéolyse, un refoulement des autres viscères, etc. ; la radiographie thoracique sert, elle, au bilan d’extension ;
- échographie abdominale et pelvienne : c’est l’examen phare car il précise le siège de la tumeur (intra- ou rétropéritonéal, rénal ou extrarénal, pelvien) et réalise le premier bilan d’extension (adénopathies, rapports vasculaires, etc.) ;
- TDM et/ou IRM : elles seront toujours réalisées pour préciser les localisations et les rapports de la tumeur. La TDM pulmonaire sera réalisée systématiquement en cas de néphroblastome, à la recherche de métastases. Le choix entre les deux techniques dépendra de :
- l’âge de l’enfant : sédation nécessaire pour IRM ;
- la prise en compte de l’exposition aux rayons X et de la disponibilité des machines ;
- la nécessité d’injection de produit de contraste ;
- la zone à explorer (canal médullaire : IRM) ;
- explorations en médecine nucléaire : elles sont réalisées avec la MIBG en cas de suspicion de neuroblastome (fig. 27.4) ou phéochromocytome, ou le 18FDG (ou TEP-TDM) en cas de suspi-cion de lymphome. Elles ont l’avantage de mettre en évidence des localisations métastatiques (moelle osseuse pour le neuroblastome, SNC et moelle osseuse pour le lymphome).
La biologie comprendra systématiquement une NFS-plaquettes, un dosage des LDH, un ionogramme sanguin et un dosage sanguin de la créatinine et de l’urée.
En fonction de l’orientation diagnostique seront réalisés :
- des catécholamines urinaires (rapportées à la créatininurie) : dopamine, acide homovanillique (HVA), acide vanylmandélique (VMA) en cas de masse rétropéritonéale ;
- dosage sanguin de l’AFP en cas de masse hépatique (hépatoblastome) ;
- dosage sanguin des hCG (choriocarcinome), de l’AFP (tumeur vitelline), des hormones gonadiques en cas de masse ovarienne ;
- myélogramme ± biopsie ostéomédullaire (BOM) en cas de neuroblastome ou lymphome ;
- analyse du LCS en cas de lymphome.
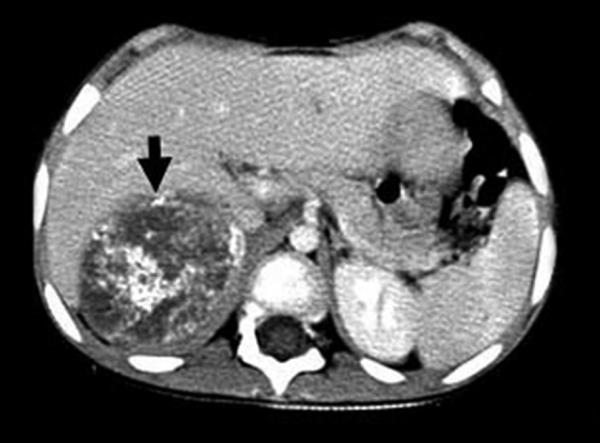
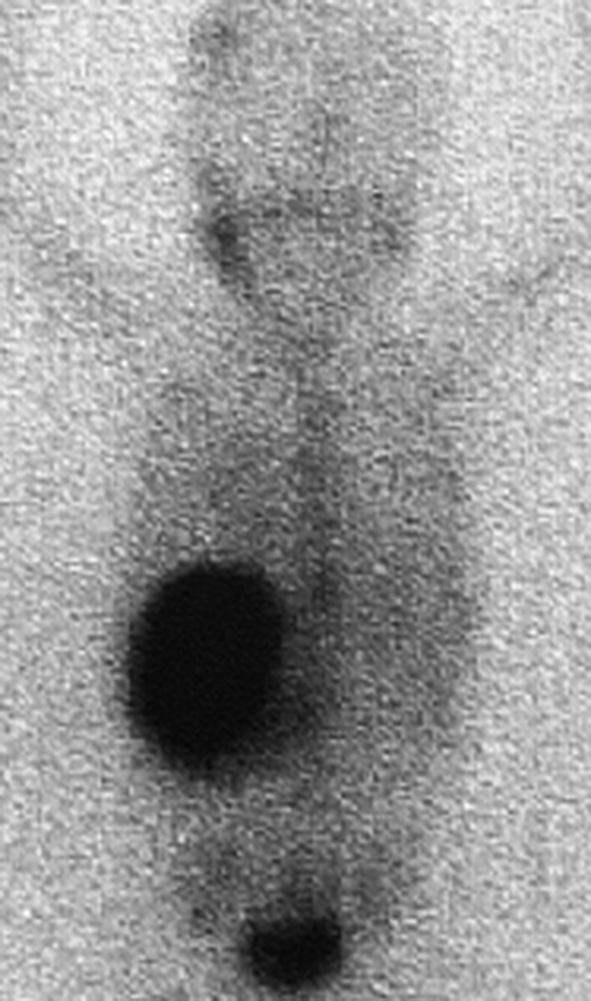
Fig. 27.4. ![]() Neuroblastome abdominal.
Neuroblastome abdominal.
A. TDM abdominale injectée, coupes axiales. Volumineuse masse surrénalienne calcifiée. B. Scintigraphie MIBG correspondante. Fixation forte de la masse et fixations rachidienne, du bassin et du crâne.
La documentation histologique et/ou cytologique est requise pour toute tumeur (à l’exception du néphroblastome si sa présentation clinique et radiologique est typique) : biopsie chirurgicale ou percutanée pour la plupart des tumeurs, ponction cytologique possible d’un épanchement pé-ritonéal en particulier pour les lymphomes, sur pièce d’exérèse pour les rares tumeurs localisées opérables d’emblée.
Ces prélèvements sont pratiqués en milieu spécialisé afin de réaliser les analyses des altérations génétiques (en particulier pour le neuroblastome à la recherche du principal facteur pronostique qu’est l’amplification du gène N-myc) et de congeler du matériel tumoral.
Au total, les neuroblastomes, néphroblastomes et lymphomes B de haut grade (Burkitt ou équivalent) représentent chacun 30 % des étiologies des tumeurs ab-dominales de l’enfant.
Le tableau 27.1 résume leurs caractéristiques principales (hors traitement).
Tableau 27.1. ![]()
![]() Caractéristiques prin-cipales des trois principales tumeurs abdominales de l’enfant.
Caractéristiques prin-cipales des trois principales tumeurs abdominales de l’enfant.
| Néphroblastome ou tumeur de Wilms |
Neuroblastome | Lymphome | |
|---|---|---|---|
| Démographie | < 5 ans | 40 % < 1 an 98 % < 6 ans |
5–10 ans Garçon |
| Signes de découverte particuliers | HTA, hématurie | HTA, compression médullaire, sites métastatiques (orbite, peau, moelle osseuse, foie) | Invagination intestinale aiguë |
| Facteur prédisposant | Wiedeman-Beckwith, WAGR, Denis-Drash | Déficit immunitaire | |
| Localisation | Rein Bilatérale possible |
Ganglions sympathiques et médullosurrénale | Péritonéale Volontiers extraganglionnaire |
| Caractéristiques de l’imagerie locorégionale | Rétropéritonéal rénal (signe de l’éperon) (fig. 27.5) Refoulement vasculaire Multilobulaire ou kystique Thrombus veineux |
Rétropéritonéal (rein refoulé) Atteinte périvasculaire infiltrante Calcifications Infiltration du canal rachidien |
Atteinte digestive multiple avec dédifférenciation de la muqueuse Atteinte ganglionnaire non mésentérique Ascite |
| Bilan diagnostique particulier | TDM pulmonaire | Catécholamines urinaires Scintigraphie MIBG Myélogramme/BOM, N-myc tumoral |
LDH (croissance très rapide) TEP-TDM Myélogramme LCS |
| Sites métastatiques principaux | Poumon | 50 % au diagnostic : peau et foie (Pepper) chez le nourrisson, péri-orbitaire, moelle osseuse | SNC, moelle osseuse |
| Pronostic | Fonction du type histologique et de l’extension tumorale Survie : 90 % avec chimiothérapie et chirurgie (± radiothérapie) |
Fonction de l’âge (< ou > 18 mois), de l’extension et de N-myc Survie très hétérogène : de < 40 % si stade IV ou N-myc amplifié à 90 % si localisé et < 18 mois |
Fonction de la présence de métastases Survie : 90 % avec chimiothérapie seule (pas de chirurgie) |
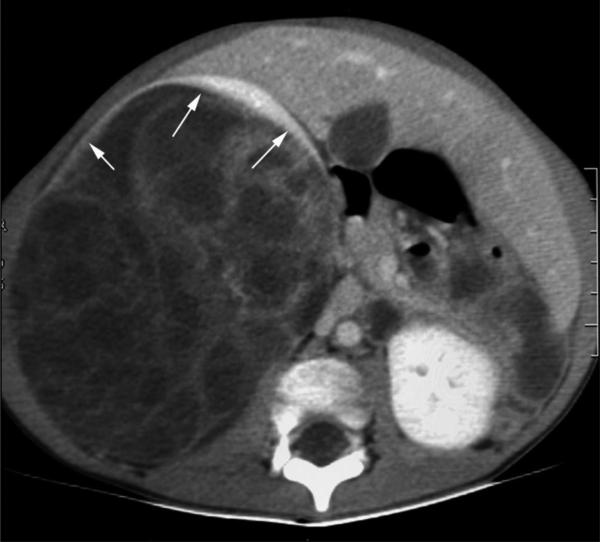
Fig. 27.5. ![]() Néphroblastome.
Néphroblastome.
TDM abdominale injectée. Coupe axiale. Volumineuse tumeur rénale droite, hétérogène, cloisonnée, prenant peu le contraste, refoulant en avant une couronne de parenchyme rénal sain (flèches blanches) raccordé à angle aigu avec la tumeur (signe de l’éperon).
D. Tumeurs osseuses
1. Généralités
Les ostéosarcomes et sarcomes d’Ewing représentent 90 % des sarcomes osseux de l’enfant et de l’adolescent. Ils sont plus fréquents chez les adolescents avec un sex-ratio H/F à 1,4/1.
Les localisations préférentielles sont les métaphyses des os longs (« près du genou et loin du coude ») pour les ostéosarcomes, tandis que les sarcomes d’Ewing touchent les diaphyses des os longs mais aussi le bassin et les côtes, ainsi que les os plats.
La prise en charge thérapeutique repose sur l’association de la chimiothérapie et de la chirurgie, voire parfois de la radiothérapie (tumeurs d’Ewing). Le pronostic des formes localisées dépend principalement de la possibilité d’effectuer une résection carcinologique de la tumeur et de l’évaluation de la réponse à la chimiothérapie sur la pièce de résection. Ces approches thérapeutiques permettent d’obtenir une survie globale de l’ordre de 60 à 75 %.
2. Diagnostic
Signes cliniques d’appel
Trop souvent, le diagnostic de tumeur squelettique maligne est retardé du fait de la méconnaissance de la symptomatologie ou de l’évocation rassurante de diagnostics bénins (entorse, douleur post-traumatique).
Devant une douleur persistante, la plus grande vigilance s’impose. Une douleur profonde localisée sur un membre, durable mais d’intensité variable, lentement progressive et secondairement d’allure inflammatoire est le principal signe d’alerte. Il faut ainsi particulièrement se méfier des douleurs réveillant l’enfant la nuit. L’état général est le plus souvent conservé.
On peut aussi retrouver :
- tuméfaction en regard de l’os atteint ;
- douleur mécanique post-traumatique (à l’occasion de la pratique du sport) ;
- fracture pathologique inaugurale ;
- inflammation locale associée à de la fièvre pouvant orienter initialement vers une infection osseuse.
Les sarcomes d’Ewing (parfois avec une composante extraosseuse importante) peuvent conduire à des signes plus aigus et trompeurs, comme des signes neurologiques par compression des racines nerveuses et/ou médullaire ou des signes de détresse respiratoire en lien avec un épanchement pleural.
Stratégie diagnostique
L’imagerie comprendra des radiographies standards qui viseront à éliminer les diagnostics différentiels et à décrire les modifications structurales de l’os : ostéolyse, réaction périostée avec un périoste décollé (aspect d’éperon de Codman, fig. 27.6A), voire spiculations transverses ou « images en feu d’herbe » (fig. 27.6B).
Le bilan radiographique sera complété par une IRM qui fournira des informations sur l’extension intraosseuse, le franchissement des cartilages de croissance, l’extension intra-articulaire et vasculo-nerveuse, la présence d’une lésion à distance sur le même os (skip métastase).
Les localisations à distance seront explorées par une TEP-TDM (ou une scintigraphie osseuse au technétium) et, de façon systématique, une TDM thoracique à la recherche de métastases pulmonaires. Une exploration ostéomédullaire sera adjointe pour les sarcomes d’Ewing (myélogramme et BOM).
La preuve diagnostique ne pourra être apportée que par l’étude anatomopathologique d’une biopsie de la tumeur. Cette biopsie est à considérer comme faisant partie de l’acte thérapeutique. Elle devra être réalisée par l’équipe qui prendra en charge le patient pour la chirurgie tumorale.
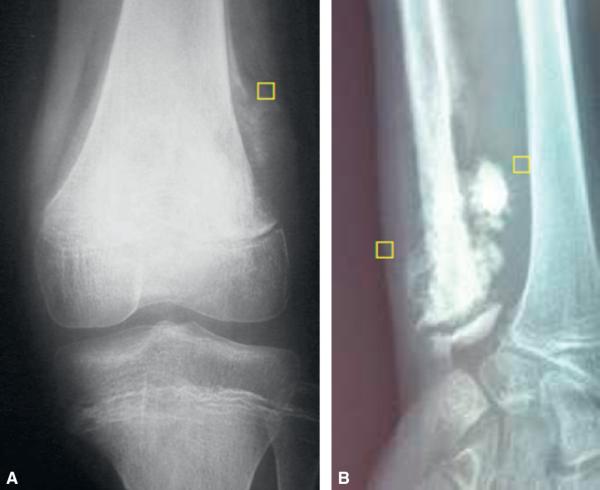
Fig. 27.6. ![]() Aspects radiographiques de tumeurs osseuses.
Aspects radiographiques de tumeurs osseuses.
A. Radiographie du genou. Éperon de Codman témoignant de l’interruption d’appositions périostées de type plurilamellaire par une tumeur agressive. B. Radiographie du poignet. Images en « feu d’herbe » témoignant d’une réaction périostée spiculée en réponse à une poussée tumorale maligne centrifuge.
Références
DH 2004 DHOS. Circulaire n° 161 du 29 mars 2004, relative à l’organisation des soins en cancérologie pédiatrique.
Lacour B, Guyot-Goubin A, Guissou S, Bellec S, Désandes E, Clavel J. Incidence des cancers de l’enfant en France : don-nées des registres pédiatriques nationaux, 2000-2004. BEH 2010;49-50:497–500.