Objectifs pédagogiques
- Expliquer les bases du conseil génétique, les possibilités de dépistage et de diagnostic prénatal (modalités et options de prise en charge dans le cadre d’une maladie d’une particulière gravité).
- Expliquer les problèmes liés à la maladie et les retentissements de l’arrivée d’un enfant souffrant de maladie génétique sur le couple et la famille.
Avant de commencer…
La mucoviscidose est la plus fréquente des maladies génétiques autosomiques récessives dans la population caucasienne. Elle est due à des mutations du gène CFTR, dont la plus fréquente en France est F508del.
Le dépistage néonatal généralisé en France depuis 2002 est réalisé à J3 de vie sur papier buvard. En l’absence de dépistage, le diagnostic postnatal est retardé, car les manifestations cliniques (principalement pulmonaires et digestives) peuvent survenir plus tardivement.
Le test de la sueur est l’examen diagnostique de référence. Il doit être complété par la mise en évidence de mutations pathogènes associées à la mucoviscidose sur chaque allèle.
La mucoviscidose est une maladie chronique d’aggravation progressive avec l’âge, qui met en jeu le pronostic vital. Le suivi multidisciplinaire est assuré dans des centres spécialisés.
La prise en charge thérapeutique repose sur une thérapie ciblée sur les symptômes et sur une thérapie corrigeant la protéine CFTR défectueuse. La prise en charge respiratoire repose avant tout sur une kinésithérapie respiratoire quotidienne et le traitement des surinfections bronchopulmonaires. La prise en charge digestive repose sur le recours aux extraits pancréatiques gastroprotégés et aux vitamines liposolubles ainsi que sur des recommandations nutritionnelles. Depuis 2012, des traitements ciblant la protéine CFTR défecteuse, appelés modulateurs de CFTR, sont disponibles pour près de 90 % des patients ; ils ont profondément amélioré le pronostic.
I. Pour bien comprendre
A. Épidémiologie
La mucoviscidose est la plus fréquente des maladies génétiques autosomiques récessives létales dans la population caucasienne.
En France, sa fréquence est de 1/4 700 naissances et la proportion des sujets hétérozygotes porteurs sains estimée à 1/34. En 2023, plus de 7 200 patients sont suivis en France avec 60 % des patients suivis dans des centres de ressources et de compétences pour la mucoviscidose (CRCM) adultes.
La survie des patients s’est considérablement améliorée puisque l’on considère que la médiane de survie d’un patient né en 2010 est de l’ordre de 50 ans.
Les récents modulateurs de CFTR vont certainement permettre d’améliorer encore plus la survie des patients mais le recul n’est pas encore suffisant en 2023 pour l’apprécier.
B. Rappels de génétique et physiopathologie
Cette affection est due à des mutations du gène CFTR (Cystic Fibrosis Transmembrane conductance Regulator), situé sur le bras long du chromosome 7, codant une protéine transmembranaire intervenant dans la régulation du transport transépithélial des ions chlorure (Cl–).
L’absence ou la dysfonction de la protéine CFTR entraîne alors un défaut du transport du Cl– et une augmentation de la réabsorption de sel et d’eau. Il en résulte notamment au niveau de l’épithélium bronchique, une réduction du liquide de surface bronchique, altérant la clairance mucociliaire.
Il s’agit donc d’une exocrinopathie généralisée, touchant les glandes séreuses et les glandes à sécrétion muqueuse qui entraîne une accumulation de sécrétions visqueuses et déshydratées. Ceci induit la formation d’un « mucus visqueux » (d’où le nom de mucoviscidose) qui obstrue différents sites de l’organisme, notamment l’appareil respiratoire, le tube digestif et ses annexes (pancréas, voies biliaires et foie), les glandes sudoripares et le tractus génital.
Plus de 2 000 mutations ont été identifiées à ce jour. Elles peuvent être soit identiques sur les deux allèles (homozygotie) soit différentes (hétérozygotie composite).
En France, la mutation la plus fréquente (environ 80 % des cas) conduit à la délétion du 508e acide aminé (phénylalanine), appelée F508del.
1/4 700 naissances, transmission autosomique récessive, hétérozygotie 1/34, gène CFTR, F508del.
II. Diagnostiquer une mucoviscidose
A. Quand évoquer le diagnostic ?
1. Diagnostic anténatal ciblé
Histoire familiale
Le diagnostic peut être évoqué d’emblée si un enfant du couple est atteint de mucoviscidose ou en cas d’identification d’un couple à risque 1/4 dans un contexte familial.
Un conseil génétique se justifie notamment dans les situations suivantes :
- antécédent de mucoviscidose chez un enfant du couple (les deux parents, sains, sont obligatoirement hétérozygotes) ;
- situation d’hétérozygotie connue chez l’un des deux parents (recherche d’une mutation fréquente chez l’autre parent) ;
- couples ayant des apparentés proches avec enfant atteint (recherche d’hétérozygotie chez les deux parents).
Absence d’histoire familiale
Le diagnostic peut être suspecté devant une anomalie échographique :
- hyperéchogénicité intestinale ;
- dilatation digestive
- non-visualisation de la vésicule biliaire ;
- images évoquant une péritonite méconiale.
La conduite diagnostique consiste à réaliser une étude génétique chez les deux parents, avec tracé de l’arbre généalogique et recherche des mutations les plus fréquentes en fonction de l’origine géographique ou ethnique des parents, après information éclairée et consentement signé de chacun des parents.
Si les deux parents sont hétérozygotes, le risque très élevé d’atteinte fœtale conduit à proposer un diagnostic prénatal.
Si aucun des deux parents n’est porteur des mutations fréquentes, le couple peut être rassuré ; le risque résiduel que le fœtus soit atteint de mucoviscidose est établi en fonction des signes échographiques et de la sensibilité de la recherche génétique dans la population d’origine des parents.
En revanche, si l’un des deux parents est hétérozygote pour une mutation fréquente, il existe un risque que l’autre parent soit porteur d’une mutation rare, ce qui justifie le recours au séquençage complet du gène chez ce dernier. Si une mutation rare est identifiée, un diagnostic prénatal est proposé. Si aucune mutation n’est identifiée, le couple peut être rassuré.
Modalités du diagnostic prénatal
Lorsque les mutations parentales sont identifiées, elles sont recherchées chez le fœtus sur une biopsie de trophoblaste dès 12 SA.
Dans la situation rare d’un premier enfant avec mucoviscidose et de mutations non identifiées, des dosages biologiques (immunoenzymes intestinales, PAL, GT, leucine aminopeptidase) par amniocentèse au terme de 18 SA apportent des arguments diagnostiques.
Enjeux éthiques
Un diagnostic anténatal de mucoviscidose peut motiver une demande d’interruption médicale de grossesse, au titre de « maladie d’une particulière gravité et incurable au moment du diagnostic ».
Comme dans toute démarche de diagnostic anténatal, les parents doivent avoir reçu une information claire, objective, complète et adaptée à leur compréhension.
Le médecin doit évaluer avec eux le rapport bénéfices/risques du diagnostic anténatal (risque de fausse couche iatrogène de 0,5 % en cas d’amniocentèse, 1 % en cas de biopsie de trophoblaste).
Les objectifs du conseil génétique sont d’aider les couples à comprendre les solutions qui s’offrent à eux en l’état actuel des connaissances médicales, de façon qu’ils puissent prendre des décisions « éclairées tout en amoindrissant le sentiment de culpabilité ou l’anxiété ». Les impératifs de soins quotidiens, d’évolution de la maladie doivent être expliqués. Les décisions concernant le fait d’avoir un enfant malade ou de mener à terme une grossesse lorsque le fœtus est atteint appartiennent aux deux parents en fonction de leur perception sur le retentissement personnel et familial de la maladie chronique. Le respect du choix est primordial.
Dans ce contexte, l’objectif d’avoir « un bébé en bonne santé » ne constitue pas une forme d’eugénisme, c’est-à-dire « une politique coercitive visant à poursuivre un objectif de procréation allant à l’encontre des droits, des libertés et des choix de l’individu » (Problèmes éthiques rencontrés en génétique médicale ; rapport de l’OMS 2001).
Diagnostic anténatal = ciblé : histoire familiale et conseil génétique, signes évocateurs échographiques.
2. Dépistage néonatal
Le dépistage néonatal pour la mucoviscidose est généralisé en France depuis 2002.
L’intérêt de ce dépistage réside dans la prise en charge précoce des manifestations pulmonaires et digestives de la mucoviscidose, qui ralentit l’évolution de la maladie et améliore son pronostic en termes de survie.
Le dépistage est réalisé à partir du sang recueilli à J3 de vie sur papier buvard , après recueil du consentement signé des parents (pour la recherche génétique éventuelle).
Il repose sur le dosage de la trypsine immunoréactive (TIR), enzyme pancréatique dont un taux élevé reflète une souffrance pancréatique. Si ce dosage est élevé, un diagnostic moléculaire à la recherche des 29 mutations du gène CFTR les plus fréquentes dans la population française est mené.
En cas de mutation identifiée sur au moins un allèle, l’enfant est convoqué au CRCM (centre de ressources et de compétences pour la mucoviscidose) régional pour confirmation diagnostique par un test de la sueur. En l’absence de mutation retrouvée, l’enfant ne sera convoqué au CRCM que si un nouveau dosage de TIR prélevé à 3 semaines reste élevé.
Ce mode de dépistage est assez sensible mais peu spécifique (4 enfants sur 5 convoqués au CRCM suite à un dépistage positif n’ont pas la mucoviscidose). Le taux de faux négatifs du dépistage (= proportion d’enfants avec mucoviscidose et non reconnus par le dépistage néonatal) est entre 3 et 3,5 %.
Dépistage néonatal = généralisé : TIR à J3 de vie + biologie moléculaire si TIR élevée.
3. Diagnostic chez l’enfant sur des manifestations cliniques évocatrices
Malgré la mise en place du dépistage, le diagnostic de mucoviscidose peut aussi être fait sur des symptômes au cours de l’enfance, voire à l’âge adulte (enfants nés avant le dépistage néonatal ou faux négatifs du dépistage néonatal).
Manifestations respiratoires et infectieuses
Elles sont responsables de 90 % de la morbidité et conditionnent le pronostic et la qualité de vie.
Les manifestations initiales ne sont pas spécifiques :
- dyspnées sifflantes à répétition ;
- encombrement bronchique persistant.
L’évolution est marquée chez l’enfant plus âgé par la bronchopathie chronique :
- avec : toux chronique et bronchorrhée permanente ; exacerbations récurrentes avec expectorations mucopurulentes, pendant lesquelles la toux se majore, les crachats se modifient (augmentation du volume et de leur viscosité), la tolérance à l’effort diminue et l’appétit s’altère ;
- et au stade évolué : dilatations des bronches (fig. 63.1) pouvant saigner (hémoptysie), distension thoracique majeure, avec emphysème et risque de pneumothorax :
- et à long terme : insuffisance respiratoire chronique avec dyspnée d’effort devenant permanente, hypoxie nocturne puis diurne, se décompensant tardivement avec une hypercapnie et une hypertension artérielle pulmonaire.
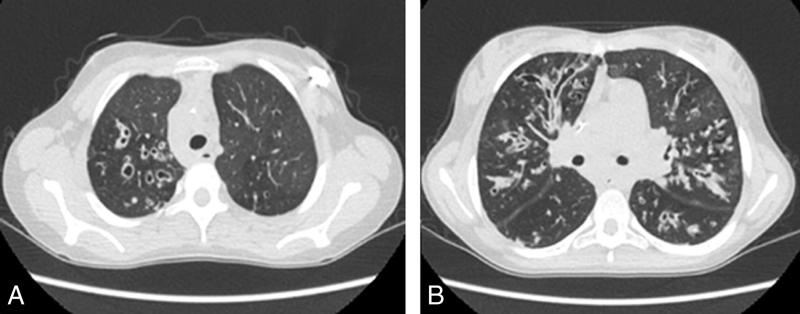
Fig. 63.1. ![]() Scanner pulmonaire d’un enfant ayant une mucoviscidose.
Scanner pulmonaire d’un enfant ayant une mucoviscidose.
A. TDM coupe axiale montrant des dilatations bronchiques du lobe supérieur droit. B. TDM coupe axiale montrant des impactions mucoïdes et des dilatations des bronches cylindriques et sacculaires.
Sur le plan fonctionnel, le syndrome obstructif s’aggrave progressivement (diminution du VEMS). Il est associé à une distension pulmonaire (augmentation du volume résiduel).
La colonisation bactérienne chronique des sécrétions bronchiques a lieu dès les premières années de vie. Il s’agit d’un signe cardinal de la maladie. Elle doit être dépistée régulièrement par examen cytobactériologique des crachats (ECBC), recueilli au cours d’une expectoration profonde ou d’un crachat induit si l’enfant n’est pas sécrétant. Les germes retrouvés initialement sont le plus souvent : H. influenzae et S. aureus sensible à la méthicilline (SAMS).
L’identification du Pseudomonas aeruginosa dans les sécrétions bronchiques peut survenir précocément. La primo-colonisation correspond au premier isolement de P. aeruginosa dans l’arbre bronchique. L’enjeu thérapeutique est l’éradication de P. aeruginosa au moment de la primo-infection ou des infections intermittentes pour éviter une colonisation chronique. La colonisation chronique est associée à un déclin plus rapide de la fonction respiratoire.
Des infections à mycobactéries atypiques (Mycobacterium abscessus ou M. avium), aspergillaires (avec des manifestations immunoallergiques, notamment dans le cadre d’une aspergillose bronchopulmonaire allergique) peuvent également survenir, comme des infections à bactéries multirésistantes : S. aureus résistant à la méthicilline (SARM), Stenotrophomonas maltophilia, Achromobacter xylososidans.
Manifestations digestives
Le nouveau-né peut présenter :
- un iléus méconial : syndrome occlusif, retard d’élimination du méconium ;
- un ictère cholestatique rétentionnel.
Le nourrisson et l’enfant associent :
- une insuffisance pancréatique exocrine (85 % des nourrissons) :
- stéatorrhée (diarrhée chronique avec selles graisseuses et nauséabondes, responsable d’une maldigestion des graisses) ;
- avec pour conséquence (si non compensée), un retard pondéral puis statural et une dénutrition, malgré une hyperphagie initiale ;
- du fait d’une malabsorption digestive, des carences en :
- vitamines liposolubles A, D, E, K ;
- oligoéléments du fait d’une malabsorption digestive ;
- une atteinte hépatobiliaire (dont l’incidence augmente avec l’âge) :
- stéatose hépatique (asymptomatique) ;
- lithiases biliaires ;
- cirrhose biliaire multifocale (5 à 15 % des patients), pouvant évoluer vers l’hypertension portale et plus rarement l’insuffisance hépatocellulaire ;
- d’autres atteintes :
- RGO ;
- prolapsus rectal ;
- constipation ± épisodes de subocclusion répétés par obstruction distale du grêle.
La dénutrition, lorsqu’elle est présente, résulte de l’inadéquation entre les besoins importants liés à l’hypercatabolisme principalement en rapport avec l’insuffisance respiratoire et les infections. Chez l’enfant non diagnostiqué, elle peut être expliquée par les pertes digestives liées à une mauvaise compensation de la maldigestion et des ingesta souvent insuffisants.
Autres manifestations
D’autres manifestations cliniques sont évocatrices mais plus rares :
- ORL : sinusite maxillaire, polypose nasale ;
- endocriniennes : insuffisance pancréatique endocrine dont l’incidence augmente avec l’âge, entraînant une intolérance glucidique puis un diabète insulinodépendant ;
- métaboliques : déshydratation aiguë hyponatrémique lors des pics de chaleur ;
- génitales : infécondité masculine par atrésie bilatérale des canaux déférents systématique chez le garçon, retard pubertaire, hypofertilité féminine ;
- osseuses : ostéopénie et ostéoporose.
Évoquer la mucoviscidose en cas de symptômes bronchiques récidivants avec cassure pondérale.
Colonisation à Pseudomonas aeruginosa = tournant évolutif péjoratif de la maladie.
B. Comment confirmer le diagnostic ?
1. Test de la sueur
Le test de la sueur constitue l’examen de confirmation diagnostique de référence.
L’anomalie fonctionnelle de la protéine CFTR se traduit au niveau de la glande sudoripare par un syndrome de perte de sel, principe de cet examen biologique.
La sueur contient normalement moins de 30 mmol/l de chlorure. Le test est pathologique si la valeur mesurée est supérieure à 60 mmol/l. Le diagnostic de mucoviscidose est affirmé après deux examens positifs.
En cas de valeurs intermédiaires (30–59 mmol/l), il faut répéter ultérieurement le test, demander une étude génétique complète du CFTR et réaliser des mesures électrophysiologiques du transport des ions chlorures en centre spécialisé pour dépister des pathologies apparentées aux anomalies du CFTR.
2. Biologie moléculaire
L’étude du gène CFTR en biologie moléculaire est le complément nécessaire du test de la sueur.
Elle a pour objectif de déterminer le génotype chez le patient : tout d’abord recherche de mutations les plus fréquentes, puis étude plus extensive si besoin.
Le sujet malade est soit homozygote pour la même mutation, soit hétérozygote composite (porteur de deux mutations différentes).
Cette étude génétique est d’autant plus nécessaire pour mettre en place les modulateurs du CFTR.
Confirmation diagnostique = test de la sueur.
III. Principes de prise en charge thérapeutique
A. Généralités
La prise en charge doit être multidisciplinaire, avec des infirmières, kinésithérapeutes, psychologues, diététiciennes, assistantes sociales, pharmaciens, médecins.
Des centres hospitaliers coordonnent ce suivi spécialisé dans le cadre des CRCM. L’organisation des soins doit se faire au maximum au domicile. La qualité et l’acceptation d’un suivi prolongé reposent sur une collaboration étroite entre l’équipe du CRCM, le médecin traitant, l’infirmière à domicile, le kinésithérapeute en ville. Elle permet la mise en place d’un suivi régulier pour une prise en charge de qualité.
L’éducation thérapeutique des parents puis de l’enfant et de l’adolescent est fondamentale, garant de l’autonomie de l’adulte.
Il est recommandé de voir l’enfant en consultation tous les 3 mois et de faire une évaluation paraclinique annuelle.
B. Traitement des principales manifestations
1. Prise en charge respiratoire
La diminution de l’encombrement des voies aériennes est assurée par :
- la kinésithérapie respiratoire, systématique ;
- pouvant être précédée d’aérosols de mucolytiques (rhDNase ou sérum salé hypertonique).
Des recommandations ont pour objectifs d’améliorer la qualité de l’environnement respiratoire : éviction du tabac, mode de garde visant à éviter au mieux les viroses respiratoires pendant la 1re année de vie, conseils d’hygiène pour éviter les réservoirs potentiellement à risque pour le P. aeruginosa.
La poursuite d’une activité physique, adaptée aux performances respiratoires est recommandée.
Une oxygénothérapie de longue durée (nocturne ou continue) est indiquée au stade de l’insuffisance respiratoire chronique. On peut y associer éventuellement une ventilation non invasive (VNI) en cas d’hypercapnie chronique.
La transplantation pulmonaire est discutée en cas d’insuffisance respiratoire chronique sévère.
2. Prise en charge anti-infectieuse
L’antibiothérapie doit être adaptée aux germes isolés sur l’ECBC.
Selon le germe visé, elle peut être orale, inhalée et/ou intraveineuse. Les cures antibiotiques orales et intraveineuses durent habituellement 2 semaines, alors que les cures antibiotiques inhalées sont souvent prolongées pendant plusieurs mois.
La mise à disposition de diffuseurs portables et de sets de perfusion prêts à l’emploi facilite la pratique ambulatoire de l’antibiothérapie intraveineuse.
Le respect du calendrier vaccinal et des vaccinations ciblées est essentiel.
Les enfants atteints de mucoviscidose doivent notamment être vaccinés contre la grippe (tous les ans), l’hépatite A et éventuellement la varicelle après 1 an (en l’absence d’antécédent clinique). La vaccination polyosidique non conjuguée 23-valente est recommandée à partir de l’âge de 2 ans en complément de la vaccination pneumococcique conjuguée.
3. Prise en charge digestive
L’opothérapie pancréatique est la compensation de l’insuffisance pancréatique exocrine par des extraits pancréatiques gastroprotégés.
Le traitement de l’atteinte hépatobiliaire fait appel à l’acide ursodésoxycholique.
4. Prise en charge nutritionnelle
Les apports énergétiques totaux doivent dans la majorité des cas être supérieurs aux apports journaliers recommandés pour un enfant sain (120–150 %). En cas de défaillance nutritionnelle, il convient de débuter une nutrition entérale.
Une supplémentation en vitamines liposolubles est indispensable (vitamines A, E, D et K), ainsi que des compléments en sodium.
En contexte d’hypersudation (été, sport, fièvre…), la déperdition sodée par la sueur nécessite d’augmenter l’apport sodé de façon systématique.
Le lait ayant un apport protidique et sodé insuffisant, il est nécessaire d’apporter du sel de manière systématique chez le nourrisson avant la diversification. L’apport adéquat peut être contrôlé par ionogramme urinaire.
5. Modulateurs de CFTR
Ces dernières années, une meilleure compréhension de la physiopathologie de la maladie a permis des avancées thérapeutiques majeures, au premier rang desquelles se situent les modulateurs de CFTR. Ciblant en particulier la protéine mutée F508del, ils améliorent considérablement la survie des patients et illustrent à quel point la mucoviscidose est l’exemple type de maladie où les avancées en recherche fondamentale conduisent à des applications thérapeutiques directes.
Lorsqu’il existe une mutation F508del, les nouvelles thérapeutiques vont avoir une triple action sur la protéine CFTR : amélioration de la maturation de la protéine et du transport intracellulaire de la protéine, amélioration de l’ouverture du canal CFTR. Actuellement l’accès à ces thérapeutiques est possible dès l’âge de 2 ans.
Ces stratégies thérapeutiques ouvrent des perspectives passionnantes et très encourageantes à l’heure où les politiques nationales de dépistage néonatal mises en place par de nombreux pays permettent une prise en charge très précoce de la maladie.
Prise en charge multidisciplinaire, CRCM.
Kinésithérapie respiratoire systématique, contrôle de l’environnement, activité physique.
Cure d’antibiothérapie adaptée aux germes de l’ECBC, vaccinations.
Extraits pancréatiques gastroprotégés, régime hypercalorique, supplémentation en vitamines liposolubles et en sodium.
Modulateurs de CFTR.
C. Autres mesures
- Éducation thérapeutique.
- Soutien psychologique.
- Aide d’associations (par exemple, Vaincre la Mucoviscidose).
- Prise en charge à 100 % par la Sécurité sociale.
- Mise en place éventuelle d’un PAI (projet d’accueil individualisé).
Références

|
Registre français de la mucoviscidose. Bilan 2021. https://www.vaincrelamuco.org/sites/default/files/registre_francais_de_… |